
Sickle cell disease (SCD) is caused by a genetic disorder characterized for a mutation of beta-globin, resulting in an abnormal hemoglobin with low solubility when deoxygenated.1 It is the most commonly found hematological disease, affecting more than 100,000 people in the United States.2 The disease represents a significant public health problem not only due to the chronicity characteristic, but, mainly, because it dues to recurrent hospitalizations for pain management, which become more frequent over the years. This generates an average annual cost of U$1,389 per patient in the USA.3 Sickle cell anemia is the most common monogenic hereditary disease in Brazil, occurring predominantly among Afro-descendants. The distribution of the S gene in Brazil is quite heterogeneous, depending on the black or Caucasian composition of the population. Thus, the prevalence of heterozygotes for Hb S is higher in the north and northeast regions (6–10%), while in the south and southeast regions the prevalence is lower (2–3%).4 There are about 50,000 cases in the country, with an incidence of 2500–3000 cases per year.5
Ischemic pain from vessel occlusion or acute painful crisis is a central clinical feature in the natural history of the disease. The pain crisis is typically nociceptive, acute, episodic and without prodromes, being mainly present in adolescents and young adults. However, there are also adult patients who have an associated neuropathic component, as already demonstrated in studies using quantitative sensitivity test (QST).6 Despite this, there is still much doubt on the mechanisms involved in the development of neuropathic pain in patients with SCD.7
The vaso-occlusive crisis (VOC) is considered the prototype of acute pain in SCD, while other conditions are associated with chronic pain: lower limb ulcers, avascular necrosis, bone infarctions, chronic osteomyelitis, osteoporosis and chronic arthropathy.7 The acute pain crisis begins with occlusion of microvascularization by sickle cells, leading to ischemia and hypoxia, triggering tissue and vascular damage. The event causes the release of inflammatory mediators such as substance P, endothelin-1 and prostaglandin E2 (PGE2) with consequent activation of nociceptors. In addition, there is activation of N-methyl-D-aspartate (NMDA) and alpha-amino-3-hydroxy-methyl-5-4-isoxazolpropionic (AMPA) receptors, which contribute to the development of central sensitization.7
The administration of opioids is a common approach to episodes of pain in SCD. A study of 3,882 patients with SCD showed that 40% of them used opioid medications over a 12-month period and that 23% of adult patients needed more than 30 mg of morphine equivalents daily.8 However, there was not necessarily a positive correlation between the use of opioids and improved quality of life, since higher doses of medication (> or = 90 mg equivalent to oral morphine) were related to worse quality of life.9 It is known that the use of high doses of opioids increases the incidence of several side effects, which causes damage to the patient's daily life.
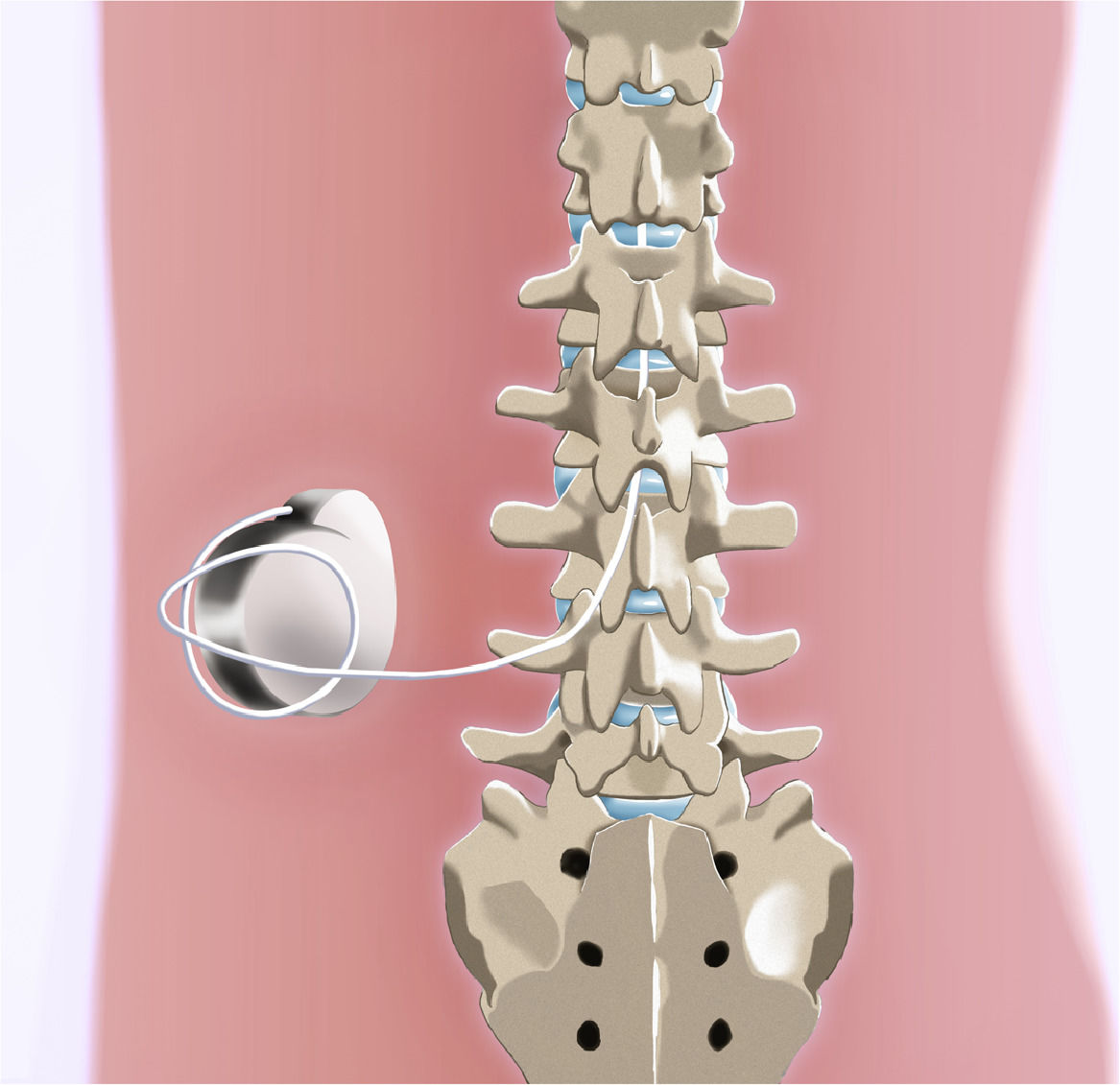
The use of intrathecal opioid delivery implantable systems consists of a well-established treatment for cancer pain and benign chronic pain in special situations.10 The intrathecal infusion system consists of a metallic device that acts as a drug reservoir, which is implanted in one of the flanks and connected to a catheter whose other end is inserted in the subarachnoid space (Figure 1). The use of this administration route reduces systemic exposure and achieves pain relief with much lower doses than oral or intravenous, causing less adverse effects to the patient.9 The purpose of the present manuscript is to describe a case of implantation of an intrathecal infusion pump for the treatment of a patient with refractory chronic pain related to SCD and to discuss therapy critically.
Case report.")
One male patient, diagnosed with SCD, evolved throughout his life with several episodes of pain. The patient's type of sickle cell disease is type SC, in which there is the presence of hemoglobin S in heterozygosis with hemoglobin C, an association defined by SC hemoglobinopathy (HbSC). He had a baseline hemoglobin of 10.5 g/dL and uses 1000 mg/day of hydroxyurea. A hemoglobin electrophoresis performed on October 2018 presented the following result: A1: 0%; A2: 4,1%; F: 9%; S: 44,3%; C: 42,6%.
At 19, he had pain in the hips, knees and, mainly, in the lumbar spine, which was persistent. This situation required several admissions to the emergency department for analgesia and hospitalizations. During crises, the patient presented relief with intravenous morphine (10mg/ml) every 4 hours, but the medication caused effects not tolerated by him.
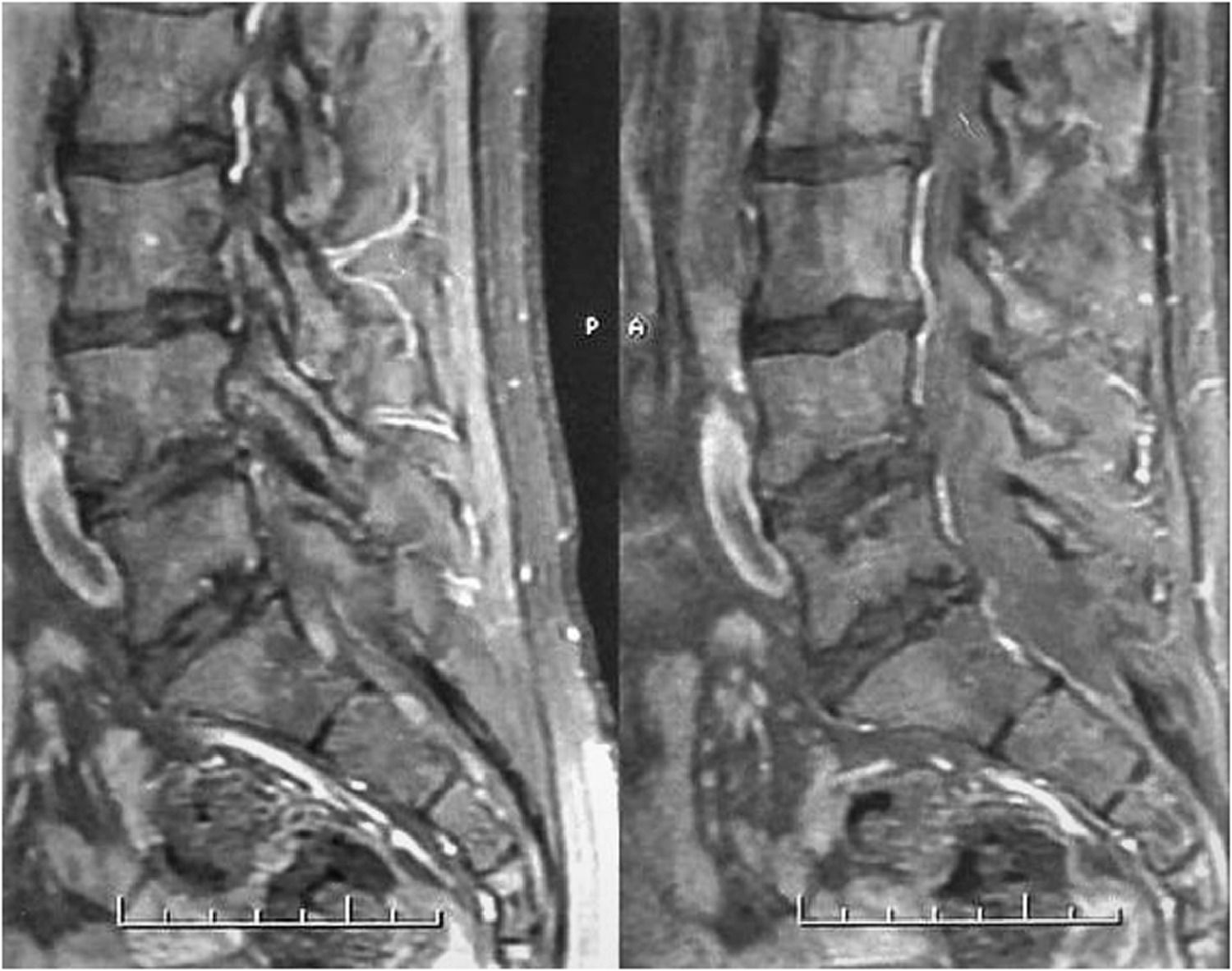
Over the years, at different times, he was treated with codeine, tramadol, methadone and oxycodone, but the analgesic outcome was insufficient. Magnetic resonance imaging of the lumbar spine showed signs of multisegmental bone infarction (Figure 2). At 29 years old, after 10 years of conservative treatment with severe pain measured by the Visual Analogue Scale (VAS) of 8/10, an intrathecal test with morphine (200mcg) was performed. The test was satisfactory, which motivated the implantation of an intrathecal infusion pump. The patient has been using the device for approximately nine years. It was replaced two years ago, due to an ulceration that appeared in the topography of the implant. Currently, he uses an electronic device with a capacity of 40 ml of medication - in this case, morphine 10 mg / ml, in a daily dose of 4705 mcg. He works as a physiotherapist and he does not need to go to the emergency department for analgesia anymore. He also does not demand analgesics orally and has mild pain (average daily VAS of 2/10).
Discussion
The management of pain in VOC is complex and requires multiple approaches. Common painkillers, non-steroidal anti-inflammatory drugs and opioids are usually administered. In addition, non-pharmacological therapies such as relaxation, hypnosis, local heat, ice and acupuncture have already been studied and are part of the therapeutic arsenal.10 Despite efforts, pain control is a major challenge and represents the most common cause of hospitalization for patients with SCD in more than 90% of cases.11
Pain control in SCD is object of research in several centers around the world, due to its recurrence and frequent hospital readmissions. In a prospective study involving 182 individuals with SCD hospitalized due to pain, it was observed that about half of the patients were readmitted to the hospital in the first month after discharge and 16% in the first week. Data analysis found that readmission in the first week after discharge was associated with increased mortality.12 Another study with a similar methodology identified that frequent and prolonged hospitalizations are risk factors for early mortality in patients with SCD.13
The fight against pain in SCD is studied on several approaches, including drugs already in use in the clinical routine such as Hydroxyurea, Pregabalin, Ketamine, as well as biological ones with blood transfusion and the development of new drugs such as crizanlizumab and of L-Glutamine.14–20 In a double-blind, placebo-controlled study, crizanlizumab (humanized anti-P-selectin monoclonal antibody) showed promise as a medication to prevent VOC, although it did not show a reduction in the rate of hospitalization days.18,19 In November 2019, this drug received FDA approval in the USA, being indicated to reduce the frequency of VOC in individuals aged ≥ 16 years and diagnosed with SCD.21 L-Glutamine, when compared to placebo, proved to be effective in reducing the number of pain attacks.19
Despite the promising potential of new pharmacological interventions aimed at changing the clinical course of VOC, opioid analgesics remain the basis for pain management. In this scenario, it is noteworthy that the use of opioids by oral route may cause intolerable adverse effects. In high doses, they can lead to an increased risk of chronic heart failure and respiratory consequences.8 They are also associated with worsening quality of life, hyperalgesia, depression and anxiety.9
One measure performed with some frequency in patients during VOC is the use of patient-controlled analgesia (PCA). In this sense, a randomized and controlled study was carried out to evaluate the effectiveness of intravenous administration of morphine through PCA systems compared to continuous infusion of the same opioid. It was observed that, in the PCA group, patients had lower cumulative mean consumption of morphine, fewer side effects, reduced length of stay, even though the pain intensity scales were similar.22 In another study, treatment with PCA was also associated with shorter hospital stay.23 This is an important finding, however, in both studies, patients triggered the pain crisis that motivated them to go to the hospital. This finding allows us to suggest that home pain control was not adequate or borderline to the development of side effects and, for this reason, it was insufficient. The reduction in hospital stay is a significant gain, but it would be even more representative if we were able to minimize hospital admission.
In 2005, Smith and colleagues published an article in which they describe good outcomes in two cases after using IDDS, one with morphine and the other with hydromorphone, for the management of refractory pain in patients with SCD.24 Six years later, in a case report of two patients with SCD who received an IDDS implant with ziconotide, MacDowell and colleagues observed good pain management, avoiding pain crises and reducing emergency room visits. Ziconotide is a direct and selective inhibitor of N-type calcium channels.25 Satisfactory results were also described in 2013 by Udeshi et al. from the use of IDDS with hydromorphone in a patient with pain crises related to sickle cell disease refractory to conventional drug treatment. The authors emphasized the possibility of additional benefit from using boluses from the Personal Therapy Manager (PTM) to help monitor and adjust patient therapy.26 Such a measure can be considered as a safe and effective therapeutic option for patients with refractory chronic pain related to SCD, since it is a method used for many years in other chronic painful contexts. Efforts must be made to ensure that health professionals involved in caring for patients with this type of clinical condition are informed about this therapeutic modality, which is often unknown, by inserting specialists in pain medicine in the management of these patients. The management of chronic refractory pain in sickle cell disease with IDDS can even be considered as a measure to prevent pain crises and thus reduce hospital admissions.
Within the therapeutic context, in a recent systematic review involving approximately 600 patients, Cooper and colleagues (2019)10 did not find consistent evidence in randomized clinical trials due to the efficacy or losses resulting from the pharmacological interventions currently used to treat VOC-related pain. Their conclusion leaves inconclusive the question about which would be the best therapy to be adopted. The approach of treating pain symptomatically with increasing doses of analgesics (opioids or not) should be considered outdated, since multimodal analgesia based on a structured, individualized therapeutic regime based on crisis prevention and reducing hospital admissions can generate better therapeutic, economic and social results.
ConclusionThe high proportion of SCD-related costs associated with hospital admissions suggests that interventions that reduce complications, such as pain crises, may also be interesting from an economic point of view. The result reported in the present manuscript must be interpreted, obviously, in the context of its limitations, since it is a purely descriptive study. However, considering the rarity of specific studies related to this therapy in this clinical context, this article opens horizons for the possibility and the need to develop prospective, controlled studies with a larger number of patients.


