
Acute myeloid leukemia (AML) is a heterogeneous disease and approximately one-third of its carriers do not have evident genetic abnormalities. The mutation of specific molecular markers, such as fms-like tyrosine kinase 3 (FTL3) internal tandem duplication (ITD), FLT3 tyrosine kinase domain (TKD) and nucleophosmin (NPM1), are associated with an adverse and favorable prognosis, respectively.
ObjectiveThe objective was to determine the prevalence of FLT3/ITD and NPM1 in Chilean patients and their association with clinical data and prognosis.
Method and ResultsTwo hundred and thirty-two children were studied between 2011 and 2017, the median being 8.6 years (ranging from 1 to 18 months). Acute promyelocytic leukemia (APL) was diagnosed in 29%. The FLT3/ITD-mutated in non-promyelocytic AML was at 10% (14/133) and the FLT3/TKD, at 3.7% (2/54). In APL, it was at 25.4% (16/63). In non-promyelocytic AML, the FLT3/ITD-mutated was associated with a high leucocyte count, the median being 28.5 x mm3 (n = 14) versus 19.4 x mm3 (n = 119), (p = 0.25), in non-mutated cases. In APL, the median was 33.6 x mm3 (n = 15) versus 2.8 x mm3 (n = 47), (p < 0.001). The five-year overall survival (OS) in non-promyelocytic AML with non-mutated and mutated FLT3/ITD were 62.7% and 21.4%, respectively, (p < 0.001); the 5-year event-free survival (EFS) were 79.5% and 50%, respectively, (p < 0.01). The five-year OS in APL with non-mutated and mutated FLT3/ITD was 84.7% and 62.5%, respectively, (p = 0.05); the 5-year EFS was 84.7% and 68.8%, respectively, (p = 0.122). The NPM1 mutation was observed in 3.2% (5/155), all non-promyelocytic AML with the normal karyotype.
ConclusionThe FLT3/ITD mutation was observed more frequently in APL and associated with a higher white cell count at diagnosis. However, the most important finding was that the FLT3/ITD mutation was associated with a shorter survival in non-promyelocytic AML.
Acute myeloid leukemia (AML) is a genetically heterogeneous disease and constitutes 20 to 25% of all childhood leukemia. The World Health Organization (WHO) 20161 defines AML according to different cytogenetic and molecular abnormalities. The most common recurrent cytogenetic abnormalities in children with AML are: t(15;17) (PML-RARA) in 10 to 12%, t(8;21) (RUNX1-RUNX1T1) in 10 to 17%, inv16 (CBFB-MYH11) in 5 to 10% and KMT2A (ex MLL) in 4 to 20%. Approximately 20 to 30% have a normal karyotype (NK).2–5
Risk stratification is generally based on genetic and molecular alterations present in leukemic blasts, in combination with early response to treatment, defined as complete remission (CR) after one or two cycles of chemotherapy.4
During the last few years, several genetic mutations have been identified that categorize AML into subtypes with different prognostic implications, specifically in those with NK. Among them are Fms-like tyrosine kinase 3 (FTL3), nucleophosmin (NPM1) and CEBPA genes.6–17 The FLT3 Internal Tandem Duplication (ITD) is the most commonly mutated gene in NK patients and in AML-M5 and it is associated with a high leukocyte count and risk of relapse and an adverse outcome.8,11 On the other hand, if the NPM1 and CEBPA are mutated and the FLT3 is not, the outcome is favorable.12,13 These patients may be included in a standard risk group and receive less intensive treatment. However, if the NPM1 is mutated and associated with the FLT3-ITD mutation, the prognosis is poor, with a greater risk of relapse14 and a need for intensive chemotherapy or consideration of administering an allogeneic stem cell transplant (allo-SCT). These proteins are posible therapeutic targets for drugs, such as sorafenib and midostaurine.15 The prevalence of the FLT3-ITD and NPM1 mutations in pediatric AML, is approximately 10 to 20%11,16 and 6 to 8%,13,17 respectively. In APL, the FTL3/ITD is mutated more commonly than in the other types of AML, 27 to 38%8–10 and is associated with a higher white cell count, M3 variant and shorter survival, mainly due to the high risk of death during induction therapy (47% versus 0%).10
The aim of this study was to determine the incidence of the FLT3/ITD, FLT3 Tyrosine kinase domain (TKD) and NMP1 mutations in children with AML in Chile. These mutations were correlated with clinical data, cytogenetics and response to treatment to optimize risk stratification in future protocols.
Material and methodsPatientsThis prospective study included all the cases of AML in children aged 18 years or less whose samples were submitted to the Molecular Biology Laboratory of the Hospital del Salvador for analysis. The study subjects presented to 17 public hospitals across Chile between January 2011 and December 2017. The study was approved by the Ethics Committee of the Metropolitan Orient Area of Santiago. Each patient or parent gave written consent to participate. The study was performed according to the Helsinski Declaration and was funded in part by a Grant from the Fundación Nuestros Hijos.
SamplesBone marrow or peripheral blood samples were obtained at diagnosis. Diagnostic analyses were centralized and conducted at the Hematology Lab, Hospital del Salvador. Analyses consisted of a morphologic review, the cases being categorized according to the French-American-British (FAB) classification system, and subsequently confirmed by flow cytometry (FACSCalibur, Becton & Dickinson, B&D), using monoclonal antibodies: anti CD2, CD7, CD11b, CD13, CD14, CD15, CD19, CD33, CD34, CD64, cCD71a, CD117, HLA-DR., MPO and anti-lisozima.
Cytogenetics and fluoresceine in situ hibridization (FISH)Cytogenetics and FISH were performed at two reference laboratories, Hospital Luis Calvo Mackenna and del Salvador, using culture and G banded, and clonal abnormality defined according to the International Nomenclature of Human Cytogenetics System.18
Molecular studiesMolecular analyses were performed at the Molecular Biology Laboratory, Hospital del Salvador, using the real-time polymerase chain reaction (RT-PCR) (Rotor Gene Q/Quiagen).
Risk stratificationPatients were stratified into 3 risk groups:15 favorable: t(15;17), t(8;21) and inv16; intermediate: NK, or other types that were neither favorable or unfavorable, including 11q23, and; unfavorable: −7, −5, or structural alterations of 5p or 5q, t(6;9), complex karyoype (≥ 3 alterations) or 12p abnormalities.3–5 Cases without cytogenetic information and with no recurrent molecular abnormalities were designated as intermediate risk.
TreatmentPatients received treatment at their referring center. Those under 15 years old received the PINDA-LMA 2006 Protocol.19 Cases under one year old received the same protocol, but with ARA-C doses adjusted for age. Those with Down Syndrome also received PINDA-LMA 2006, but with a reduced antracycline dose. Patients with Acute Promyelocytic Leukemia (APL) received the Internacional APL Study protocol. Patients between 15 and 18 years of age, received the PANDA-LMA protocol20 and those with APL were treated using the LPA 2000 Protocol. Patients who did not acheive complete remission (CR) during induction were treated with HAM (citarabine 3 g/m2 every 12 h/day iv for 3 days and mitoxantrone 10 mg/m2/day iv days 3 and 4). The allogeneic stem cell transplant (all-SCT) was performed at the Hospital Luis Calvo Mackenna.
Response to treatmentComplete remission (CR) was defined as < 5% blasts and no extramedullary disease. Refractory disease was defined as > 15% blasts post-induction or post-reinduction. Overall survival (OS) was defined as the date of diagnosis to date of death from any cause and event-free suvival (EFS), as the date of diagnosis to the first event (relapse or death from any cause without relapse).
Prognostic factorsNon-promyelocytic AML and APL were evaluated separately, with respect to the prognostic significance of the FLT3/ITD and NPHM1 and their correlation to the white cell count (WCC) at diagnosis, risk of relapse and risk of death.
Statistical analysisContinuous variables were expressed as mean (±standard deviation) if they were distributed normally, or otherwise, as median (percentile 25 - 75). The Student's t-test was used to evaluate quantitative variables with normal distribution and the Mann-Whitney U test was used for variables that were not normally distributed. For categorical variables, the data were expresed as a percentage and the independent test based on χ² was used to determine significant differences.
To evaluate the correlation between the OS or EFS and FLT3/ITD positivity, the logistic regression model was used, with adjustment for univariate variables, such as age, sex, WCC at diagnosis and normal karyotype. The association between the FLT3/ITD mutated or non-mutated with the OS and EFS was reported as an Odds ratio (OR) and was considered significant if the 95% Confidence Interval (CI95%) did not exceed 1. Kaplan-Meier survival curves were generated. A p-value of < 0.05 was considered to be statistically significant. The data were analyzed using the STATA software (serial no. 301,406,367,260).
ResultsTwo hundred and thirty-two AML cases, for which samples were available in the Molecular Biology Lab, were analyzed. Eighteen children were known to have Down's Syndrome. Four cases with the myelodysplastic syndrome with excess blasts were excluded.
Patients characteristicsClinical characteristics are shown in Table 1. As a whole, 71% (164/232) were non- promyelocytic AML and 29% (68/232), APL. Cytogenetic information was obtained for 207 cases (89%) and the karyotype was normal in 49 (24%). Cytogenetic alterations are presented in Table 2. Risk stratification showed that the percentage of patients with low and intermediate risk was similar and that only 4% fell into the high-risk category. Six out of 24 cases without cytogenetic data were classified according to recurrent molecular alterations and the rest, with no informative cytogenetic/molecular information, were allocated to the intermediate-risk group.
Characteristics of 232 children with acute myeloid leukemia.
M/F: male/female; CNS: central nervous system.
Cytogenetic alterations in 207 children with acute myeloid leukemia.
All patients, except three who died at diagnosis, were treated. A hundred and ninety-five of 229 cases (85%) achieved CR, which in three cases was after reinduction with HAM. Twenty patients proceeded to all-SCT: seven low-risk (in the second CR), 12 intermediate-risk and 1 high-risk (in the first CR). Fifty-five patients died of leukemia, 32 due to treatment complications and four during transplant, and one patient committed suicide.
Prevalence and characteristics of FLT3/ITD mutationThe clinical and laboratory characteristics of non-APL AML and ALP patients with mutated and unmutated FLT3 are shown in Tables 3 and 4.
Characteristics of patients with non-promyelocytic AML with mutated and non-mutated FLT3/ITD (n = 133).
M/F: male/female, NK: Normal karyotype, CR: complete remission.
Characteristics of patients with promyelocytic AML with mutated and non-mutated FLT3/ITD (n = 63).
M/F: male/female, CR: complete remission.
The FLT3/ITD mutation was detected in 10% (14/133) of non-promyelocitic AML and 25.4% (16/63) of APL cases (p < 0.007). The FLT3/TKD was studied in only 66 cases and was mutated in 3% of all those with non-promyelocitic AML. The FLT3/ITD was mutated in 21% (8/38) of the patients with NK, excluding children with Down's Syndrome. The FLT3/TKD could not be detected in 18 children.
There was an association between the FLT3 mutation and FAB classification, as it was present in every microgranular (M3v) subtype case (Table 4).
In multivariate analysis, the FLT3/ITD mutated was a significant predictor of outcome, with ORs of 5.1 and 3.7 for risk of death and risk of relapse, respectively, compared to those not mutated.
On the other hand, in the multivariate analysis of APL, none of the factors, including the FLT3/ITD, were predictive of death and, in the univariate analysis, only the WCC at diagnosis was an adverse factor.
Prevalence and characteristics of NPM1 mutationThe NPM1 mutation was observed in 3.2% (5/155) of the cases (median age 14), all of whom had non-promyelocytic AML and an NK. Three also had FLT3/ITD mutated and only one of these is alive. One relapsed and the other died due to SCT complications. There were two NPM1 mutated FLT3/ITD non-mutated patients, of whom one is alive and the other died during induction.
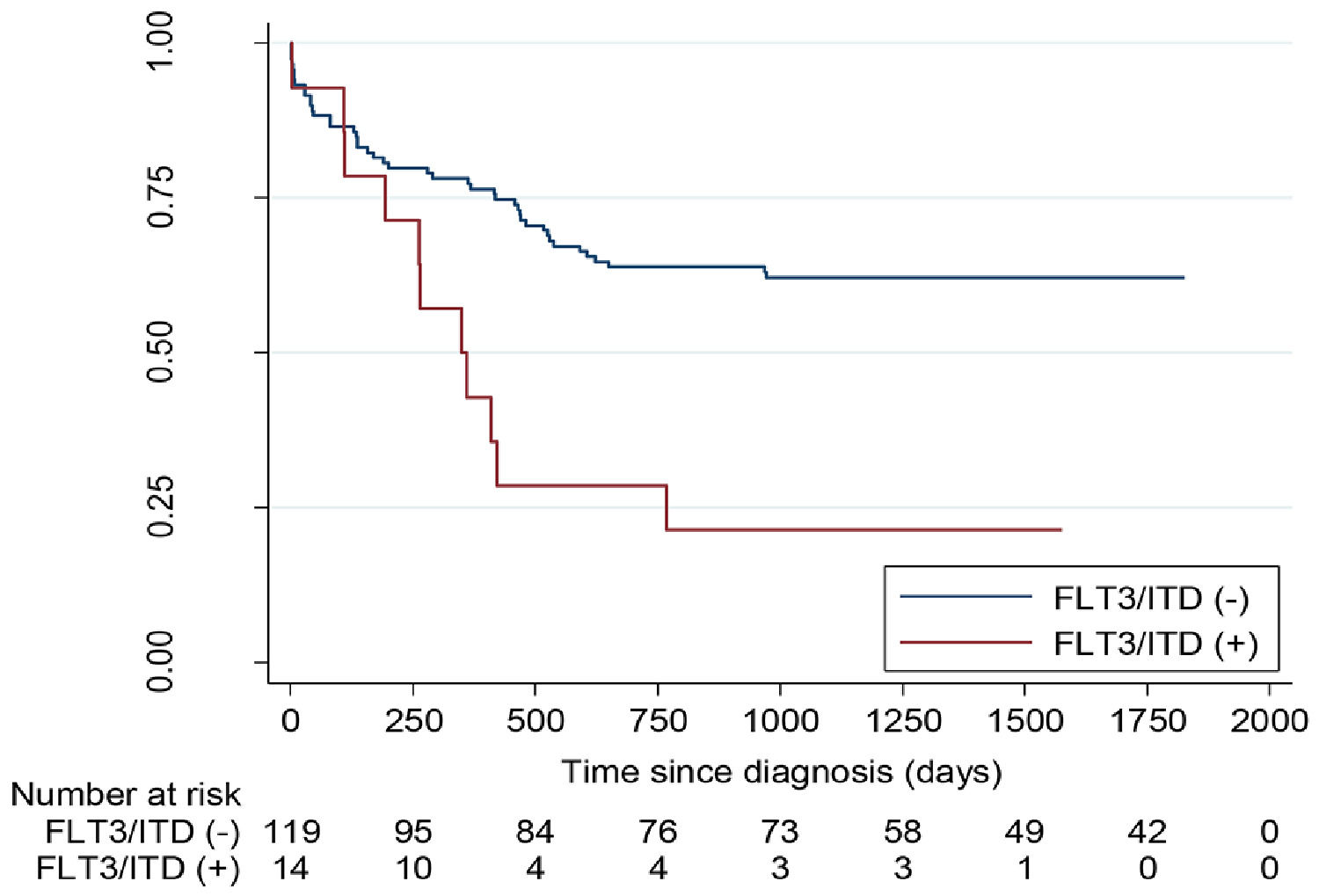
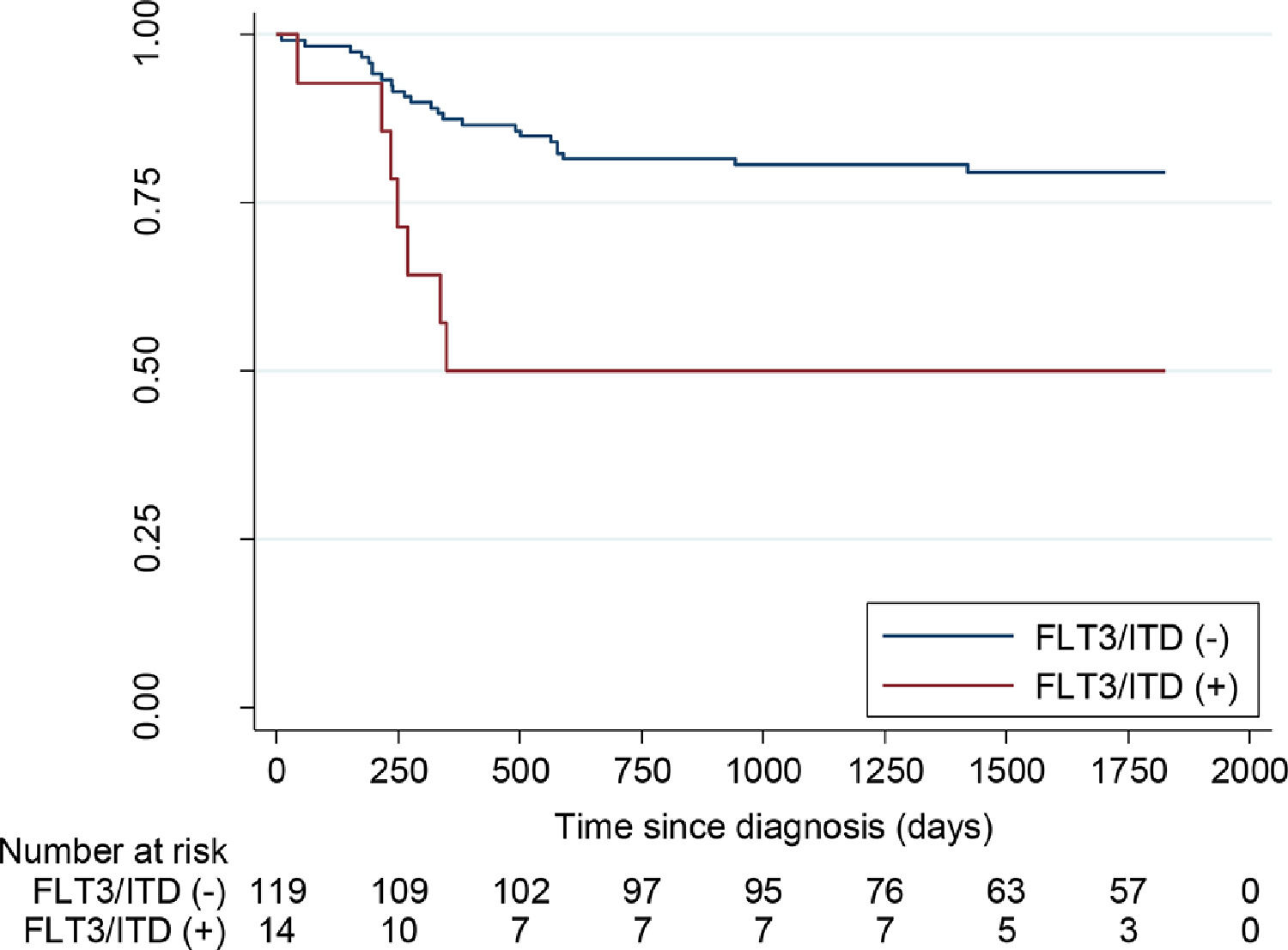
Outcome and prognosisThe CR rate was similar for the FLT3 mutated and non-mutated leukemia. (Tables 3 and 4). With a median follow-up of 35.3 months (0 - 116), the 5-year OS for children with non-promyelocytic AML, FLT3/ITD non-mutated vs mutated, was 62.7% (CI95%: 53.3 −70.7%) vs. 21.4% (CI95%: 5.2 - 44.8%) (p < 0.001) (Figure 1). The five-year EFS for patients with FLT3/ITD non-mutated or mutated was 79.5% (CI95%: 70.9- 85.8%) vs. 50% (CI95%: 22.9–72.2%), respectively (p < 0.01) (Figure 2). However, survival differences in APL for non-mutated and mutated cases did not reach significance.


The prevalence of the FLT3/ITD mutated in non-promyelocytic childhood AML and APL was 10% and 25.4%, respectively. These results were similar to those reported in the literature (12% - 16% and 27% - 31%, respectively).8–11 This mutation was also detected in approximately one-fifth (21%) of the patients with NK, although this percentage was lower than those reported by others (31%).8
It was noteworthy that the outcome of the patients with FLT3/ITD in non-promyelocytic AML was poor, due to a higher risk of relapse. This finding is concordant with the published literature,.6,7,11,16,20,21 For this reason, this genetic abnormality is now considered to be included within an adverse risk group.22
Conversely, although the FTL3/ITD mutation is more common in APL, it does not carry an adverse outcome in this type of leukemia. In a systematic review and meta-analysis that included 11 studies with 1063 patients, children and adults with APL,23 the incidence of the FLT3/ITD mutation varied from 12% to 38%. In 9 out of 11 studies, it was associated with a high WCC at diagnosis, which is known to be an adverse risk factor, compared to individuals without the mutation. Our study also found this association for APL and, in addition, for the microgranular morphology (M3v) subtype. The association was also described in 6 studies of the meta-analysis. Our study did not find an association between the FTL3/ITD mutation and death during induction or a shorter survival. One explanation could be the rather small number of cases. In a study of 104 children with APL, Kutny et al.,10 in the Children's Oncology Group (COG), described an association between the FTL3/ITD mutated and a high WCC, microgranular subtype and a higher risk of death during induction, compared to the FTL3/ITD non-mutated (47% vs. 0%, p = 0.005). Surviving induction produced no effect. In our study, the CR was achieved in 93.3% of 15 children with APL and FLT3 positive who received treatment. One patient died of CNS bleeding during the induction.
There are 3 relevant publications from Latin America,16,21,24 from Mexico, Argentina and Brazil that included 80, 216 and 703 children, respectively, solely concerning AML and not including APL. The incidence of the FLT3/ITD plus FLT3/TKD was 13% in Chile, lower than those found in Mexico (24%), Argentina (18%) and Brazil (28%). As in our study, they also determined that the FLT3 mutation was an adverse factor.
The FLT3/TKD mutation was not further analyzed, as only a small number of cases were identified.
The frequency of the NPM1 mutation at 3.2%, was lower than that reported in the literature, possibly because this group was very small.13,17,25 As in previous reports,17 we found that the NPM1 mutation was only detected in older children with NK. It is important to note that, of the three children who carried both the FLT3 and NPM1 mutations, two died, which suggests that this is a high-risk combination, as reported in the literature.22
It is important to note that this study was only possible because of the existence of reference laboratories in the Chilean Public Health System that perform a centralized service of high-quality diagnostic assays for acute leukemia in children and adults. These analyses include flow cytometry, cytogenetics and molecular biology studies. These facilities serve over 80% of the public hospitals in the country and they are a strength of the study.
In conclusion, our study shows that the FLT3/ITD mutation carries a higher risk of relapse and is therefore an adverse factor for survival in non-promyelocytic AML. In APL, the FLT3/ITD mutation was associated with a non-significant trend toward a shorter survival. The presence of this mutation may therefore provide an opportunity to combine targeted therapy with standard chemotherapy.


