
Gaucher disease (GD) is a metabolic disorder of lysosomal deposit of genetic origin, with an autosomal recessive inheritance pattern, produced by a deficiency of the acid glucocerebrosidase (GBA) enzyme.1,2
The consequence of the enzymatic defect is a lysosomal storage disease, given by the accumulation of cerebroside that occurs in the phagocytic cells of the whole organism, mainly in the cells of the reticulo-endothelial system and in some variants of the disease in the central nervous system.1,3 The approximate incidence of GD in the general population is 1/100,000 inhabitants and among Jews of Ashkenazi origin, the population with the highest incidence, is 1/500–1/1000 habitants, with 1 in 10 carriers.1,4,5 Up to April 2010, 5828 patients from all over the world were reported, of which 911 (15.6%) were reported by the Latin American Group for Gaucher Disease (GLAEG).5 There are few reports of cases in Cuba about GD, because of the low incidence of this illness in the country.2,6,7
Case reportAn 18-year-old African-American female patient was admitted at the Arnaldo Milian Hospital with the chief complaint of abdominal pain in right upper quadrant, that started two weeks ago, of moderate intensity, which was relieved slightly with the use of oral non-steroidal anti-inflammatory (NSAIDs) medication, but over time the pain stopped responding to this group of medicines. Through the physical exam we detected conjunctival pallor, and painful hepatomegaly 5 centimeters (cm) below the right costal margin. On the percussion of the left upper quadrant we were able to detect an area of dullness 8cm below the left costal margin that moved with breathing. For this presentation she was admitted to our hospital.
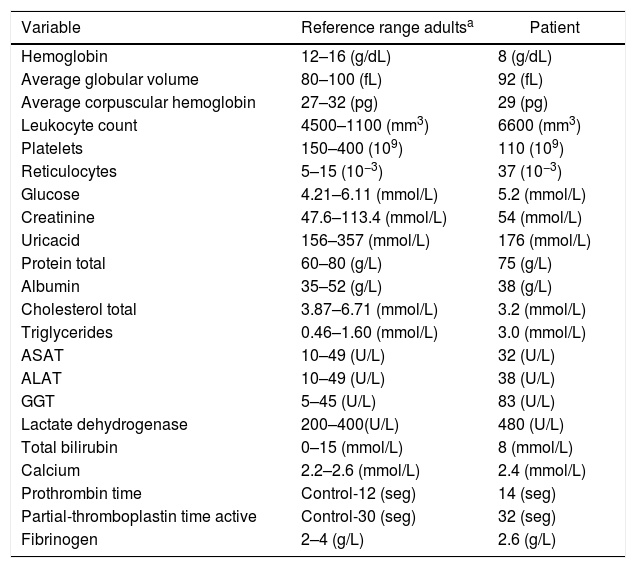
Laboratory data
| Variable | Reference range adultsa | Patient |
|---|---|---|
| Hemoglobin | 12–16 (g/dL) | 8 (g/dL) |
| Average globular volume | 80–100 (fL) | 92 (fL) |
| Average corpuscular hemoglobin | 27–32 (pg) | 29 (pg) |
| Leukocyte count | 4500–1100 (mm3) | 6600 (mm3) |
| Platelets | 150–400 (109) | 110 (109) |
| Reticulocytes | 5–15 (10−3) | 37 (10−3) |
| Glucose | 4.21–6.11 (mmol/L) | 5.2 (mmol/L) |
| Creatinine | 47.6–113.4 (mmol/L) | 54 (mmol/L) |
| Uricacid | 156–357 (mmol/L) | 176 (mmol/L) |
| Protein total | 60–80 (g/L) | 75 (g/L) |
| Albumin | 35–52 (g/L) | 38 (g/L) |
| Cholesterol total | 3.87–6.71 (mmol/L) | 3.2 (mmol/L) |
| Triglycerides | 0.46–1.60 (mmol/L) | 3.0 (mmol/L) |
| ASAT | 10–49 (U/L) | 32 (U/L) |
| ALAT | 10–49 (U/L) | 38 (U/L) |
| GGT | 5–45 (U/L) | 83 (U/L) |
| Lactate dehydrogenase | 200–400(U/L) | 480 (U/L) |
| Total bilirubin | 0–15 (mmol/L) | 8 (mmol/L) |
| Calcium | 2.2–2.6 (mmol/L) | 2.4 (mmol/L) |
| Prothrombin time | Control-12 (seg) | 14 (seg) |
| Partial-thromboplastin time active | Control-30 (seg) | 32 (seg) |
| Fibrinogen | 2–4 (g/L) | 2.6 (g/L) |
Other lab tests:
HIV (rapid test): negative
Serology VDRL: negative
Surface Ag for hepatitis B virus: negative
Antibodies to hepatitis C virus: negative
Hbelectrophoresis: AA
Fetal Hb determination: 1%.
Stool microscopy: normal microbiome
Sputum acid-fast Smear: D-cod 0 (negative)
Sputum culture: no growth of pathological organisms, normal flora
Abdominal ultrasound results: Marked enlargement of the spleen of approximately 20.1×8cm, without nodules. Liver over 6cm below the costal margin, with a homogeneous ecogenicity, no nodules. No enlarged abdominal lymph nodes. Small amount of free fluidin the peritoneal cavitybordering intestinal loops of normal diameter. Remaining organs without alterations.
Having identified hepatomegaly and giant splenomegaly, associated with thrombocytopenia and anemia, it was decided to perform a medullogram and a bone marrow biopsy.
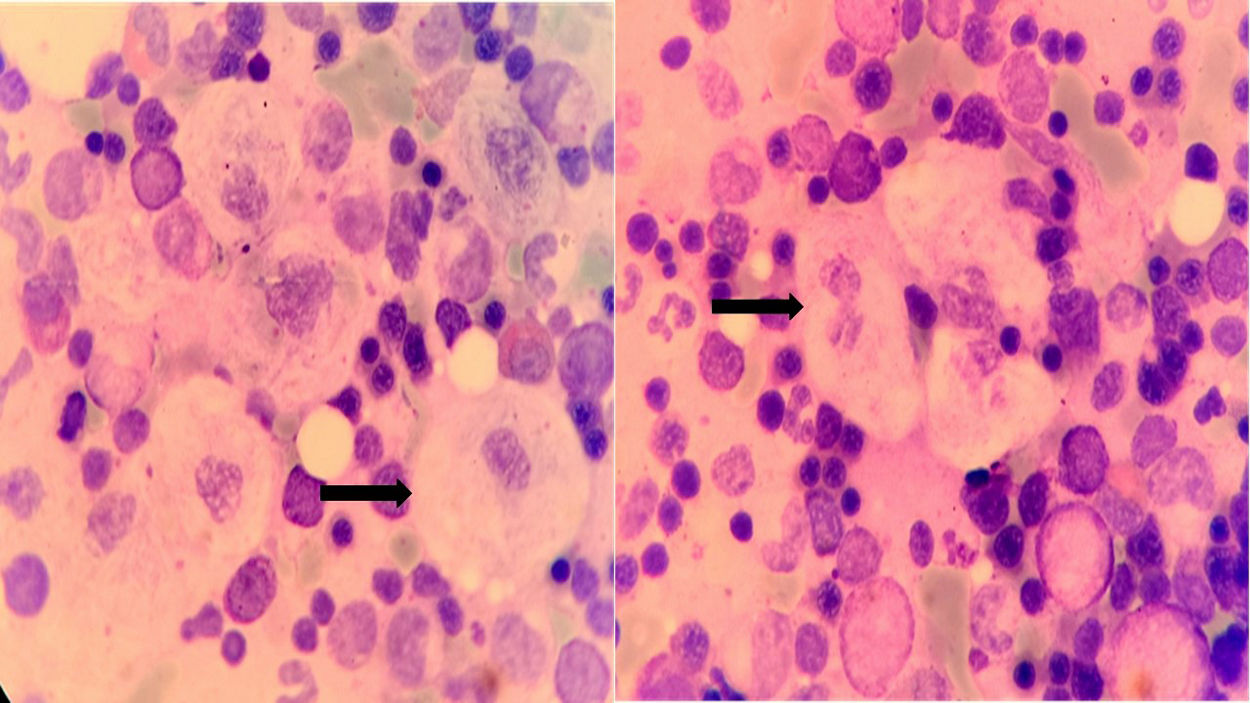
Bone Marrow Aspirate Results: Hypercellular bone marrow, with megakaryopoietic and erythropoietic hyperplasia, granulopoieticintegrity. Increase in eosinophil precursors, focal increase of mature, well-differentiated lymphocytes, increase in plasma cells below 10%. Abundant histiocytes with clear cytoplasm reminiscent of the Gaucher cell, without evidence of hemophagocytosis (Figure 1).
.")
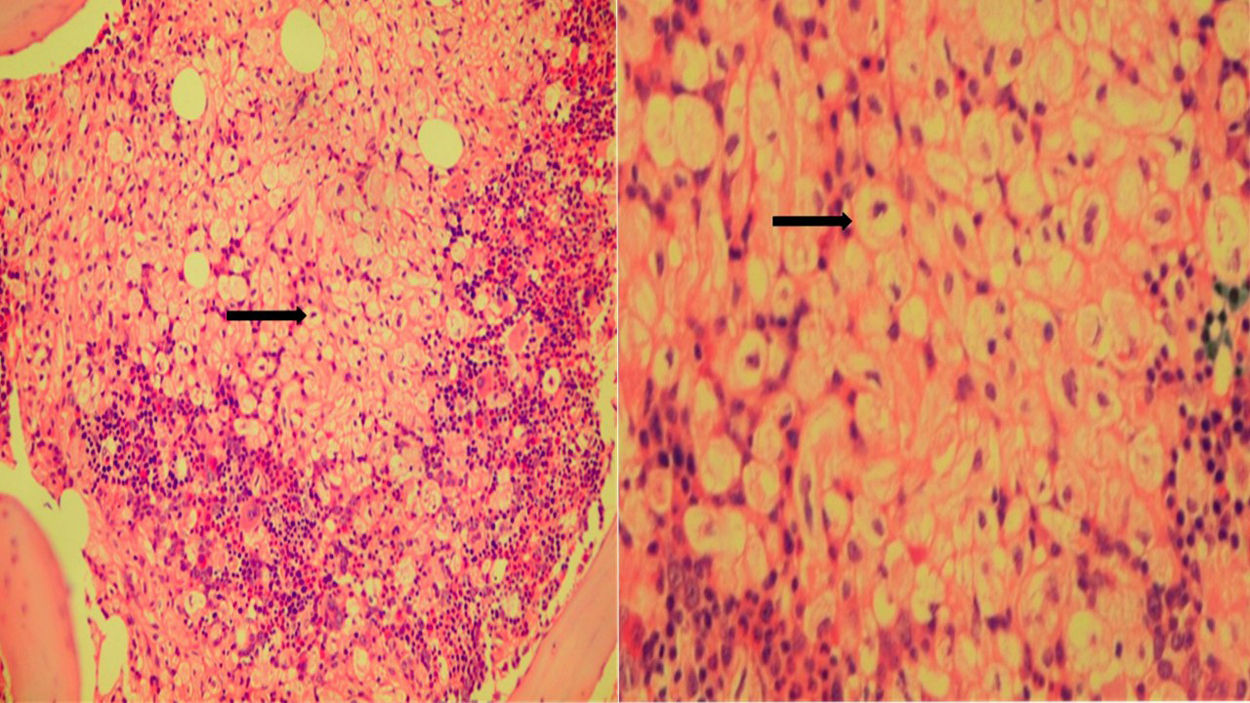
Bone marrow biopsy: Useful bone marrow cylinder, with more than 6 medullary spaces, with histiocytic cells with abundant clear cytoplasm in vacuoles and masked by remaining hematopoietic tissue. The appearance suggests a lysosomal storage disease (Figure 2).
 Bone marrow biopsy showing massive infiltration by Gaucher cells, surrounded by residual hematopoietic tissue. (B) Gaucher cells in bone marrow biopsy.")
Following the biopsy result, leukocyte glucocerebrosidase enzyme activity was determined, and found to be at 0.020nmol/mg/h (normal: 0.12nmol/mg/h). Next, the molecular gene study showed double heterozygosity for the L444P and N370S mutation, confirming the diagnosis of Gaucher disease.
DiscussionGD is characterized by its clinical polymorphism. It affects multiple organ systems and does not always correlate exactly with the specific genotype diagnosed.2
Depending on the presence or absence of neurological involvement and its severity, GD has traditionally been classified into three types.4,8
- •
Type 1 (MIM 230800): non-neuropathic form (chronic or adult form)
- •
Type 2 (MIM 230900): neuropathic form (acute or infantile form)
- •
Type 3 (MIM 231000): neuropathic form (subacute or juvenile form)
Type 1 GD is generally the most reported worldwide, However, in the largest study carried out in our population, which involves a series of 7 cases, there was a slight predominance of type 3 disease, with a total of 3 patients with type 1 disease and 4 patients with type 4 disease. There is no patient with type 2 disease.7 Type 1, by definition, does not present with associated neurological symptoms. However, recently the association of Type 1 and the so-called synucleinopathies has been described. These include peripheral neuropathies and atypical parkinsonian signs, which require regular neurological examination of patients with Type 1GD.2,4,5 Type 1 has the best survival rate of GD, and the onset of symptoms can be at any age, most often between childhood and late adulthood.1,4 In our country, the age range at the time of diagnosis has been between 4 and 52 years. In general, the main clinical manifestations include splenic and skeletal involvement, which is why its diagnosis should be considered in any child or adult who presents with hepatosplenomegaly and unexplained cytopenia.1,9 Type 1 GD is characterized by painless hepatosplenomegaly, which often leads to massive abdominal distension, as well as anemia and thrombocytopenia which can manifest as fatigue, nasal hemorrhages and easy onset hematomas. Some patients are transfusion dependent. The cytopenias are secondary to hypersplenism and infiltration of the bone marrow by Gaucher cells.1,9,10
In most patients, GD is diagnosed by hematologists, usually by means of a bone marrow trephine biopsy.1,9 Gaucher cells are usually large and can measure up to 100 microns in diameter; they may have one or more dark eccentric nuclei and rarely have vacuoles in their cytoplasm. With the electron microscope it is understood that the striated aspect of the cytoplasm is due to elongated lysosomes loaded with lipids that are deposited in double layer forming pilas.4 The differential diagnosis of “foamy” macrophages in bone marrow should be performed with lysosomal storage disorders such as: Gaucher disease, Fabry disease, Gangliosidosis GM1, Wolman/cholesterol ester, Niemanne Pick A and B (dark blue histiocytes) and hematologic disorders with pseudo-Gaucher cells such as: multiple myeloma, Hodgkin's lymphoma, non-Hodgkin's lymphoma, chronic myeloid leukemia, myelodysplasia, B-cell acute lymphoblastic leukemia, thalassemia, and sickle cell disease.9
When GD is suspected, it can be achieved by measuring the beta-glucocerebrosidase activity in the blood leukocytes, which can help avoid invasive procedures. The diagnosis must always be confirmed by a molecular study.1
More than 300 different mutations for GD have been documented by the Human Gene Mutation Database. The most common mutation in the ICGC registry is due to N370S substitution in the alleles. The second and third most common mutations are due to the substitution of L444P and the substitution of 84 GG, respectively. This can result in patients being affected by both mutations simultaneously, which is called compound heterozygosity. Mutations resulting from the N370S and L444P substitutions account for approximately 70 percent of the mutations in non-Ashkenazi European patients.1,4 A study carried out in our country with the aim of achieving molecular characterization of Cuban patients with Gaucher's disease determined the N370S and L444P substitutions as the most frequent.1,6,10
The L444P mutation is the result of a thymine forcytosine substitution in nucleotide 6433 of the active glucocerebrosidase gene that creates a new site for the restriction enzyme Nci I and causes a substitution of proline by leucine. This substitution was first described in a patient with Type 2 GD and then found in all clinical types of this disease. This is the most frequent mutation identified in patients with neurological symptoms of Gaucher's disease, where it is present in about 50% of the mutated alleles.8 However, no neurological manifestation was found in our patient.4,10
Enzyme-directed macrophage replacement therapy (TRE) has long been the standard treatment. It is not a cure for GD, because it does not repair the underlying genetic defect but it can reverse and prevent numerous manifestations of type 1 GD.7,11 In our country, the experience with this therapy is minimal due to the infrequent presentation of the disease, however the cases reported have received enzyme replacement therapy. Our patient, at the moment of this publication has so far been managed following these general measures: Red blood cell transfusion, and stimulation of granulocytic colonies with Hebervital according to requirements, which has kept the cell counts of leukocytes and hemoglobin at adequate levels. A 25% reduction of the longitudinal axis of the spleen was obtained 3 months after the start of TRE therapy.
The present case is of relevant interest to the scientific community due to its low incidence, which makes it a rarely-suspected diagnostic possibility, leading, not infrequently, to a diagnostic delay, which makes it important for medical personnel in charge of its diagnosis and monitoring.
Conflicts of interestThe authors declare no conflicts of interest.


