
The hemolytic uremic syndrome (HUS) is a syndrome of uncontrolled complement activation, associated with a high mortality rate, and often progresses to end-stage renal disease. It is clinically characterized by microangiopathic hemolytic anemia, thrombocytopenia and variable degrees of renal impairment. Diarrheal-associated HUS accounts for most of the cases. The majority of HUS cases are caused by Shiga toxin–producing bacteria, when the Shiga-toxin mediates the endothelial damage of the renal vessels and subsequent activation of the coagulation cascade.1 Another form of HUS that is not associated with Shiga or Shiga-like toxins is referred to as atypical HUS (aHUS). The aHUS accounts for 10% of the HUS cases and generally has a poor prognosis. The aHUS occurs most commonly secondary to complement dysregulation. As the complement system is an integral part of the immune system, it is constitutively regulated to prevent overactivation and downstream tissue damage. Genetic complement abnormalities have been found in up to 50% of the aHUS cases.2
Case presentationPatient 1 is a 33-year-old Caucasian man who presented to the emergency department with a 1-day history of hematuria and 3 days of fever, non-productive cough, rhinorrhea, sore throat and myalgias. He denied abdominal pain or recent history of diarrheal illness. Upon initial evaluation, vital signs were stable and the physical exam was unremarkable for any signs of superficial bleeding. Initial evaluation revealed significant acute kidney injury, indirect hyperbilirubinemia, thrombocytopenia and normocytic anemia. Further laboratory studies were consistent with intravascular hemolysis (Table 1). He reportedly had a remote past medical history of hemolytic uremic syndrome (HUS) at the age of 2, which was not obviously provoked by any illness and resolved with supportive care. Peripheral smear showed normocytic/normochromic anemia, occasional schistocytes and severe thrombocytopenia. Flow cytometric analysis of glycosylphosphatidylinositol (GPI)-linked antibodies on red blood cells, monocytes and granulocytes did not reveal any evidence of a paroxysmal nocturnal hemoglobinuria (PNH) clones. He was started empirically on plasmapheresis, given the concern regarding thrombotic thrombocytopenic purpura (TTP). Due to worsening renal function, renal biopsy was obtained, which showed thrombotic microangiopathy and acute tubular injury, with no significant tubulointerstitial scarring.
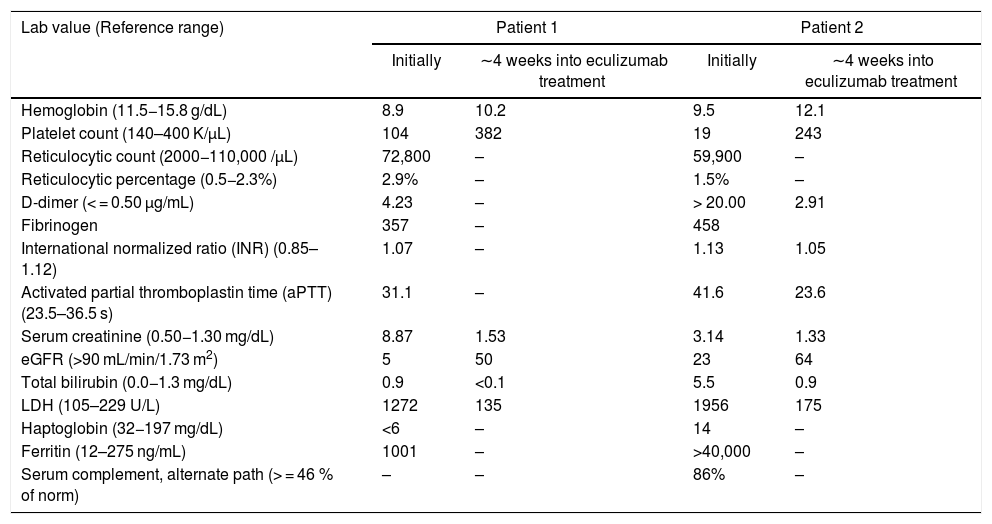
Laboratory test values on admission and after treatment initiation.
| Lab value (Reference range) | Patient 1 | Patient 2 | ||
|---|---|---|---|---|
| Initially | ∼4 weeks into eculizumab treatment | Initially | ∼4 weeks into eculizumab treatment | |
| Hemoglobin (11.5−15.8 g/dL) | 8.9 | 10.2 | 9.5 | 12.1 |
| Platelet count (140–400 K/µL) | 104 | 382 | 19 | 243 |
| Reticulocytic count (2000−110,000 /µL) | 72,800 | – | 59,900 | – |
| Reticulocytic percentage (0.5−2.3%) | 2.9% | – | 1.5% | – |
| D-dimer (< = 0.50 µg/mL) | 4.23 | – | > 20.00 | 2.91 |
| Fibrinogen | 357 | – | 458 | |
| International normalized ratio (INR) (0.85–1.12) | 1.07 | – | 1.13 | 1.05 |
| Activated partial thromboplastin time (aPTT) (23.5–36.5 s) | 31.1 | – | 41.6 | 23.6 |
| Serum creatinine (0.50−1.30 mg/dL) | 8.87 | 1.53 | 3.14 | 1.33 |
| eGFR (>90 mL/min/1.73 m2) | 5 | 50 | 23 | 64 |
| Total bilirubin (0.0−1.3 mg/dL) | 0.9 | <0.1 | 5.5 | 0.9 |
| LDH (105–229 U/L) | 1272 | 135 | 1956 | 175 |
| Haptoglobin (32−197 mg/dL) | <6 | – | 14 | – |
| Ferritin (12–275 ng/mL) | 1001 | – | >40,000 | – |
| Serum complement, alternate path (> = 46 % of norm) | – | – | 86% | – |
The ADAMTS13 (a disintegrin and metalloproteinase with a thrombospondin type 1 motif, member 13) activity was >100%, arguing against a diagnosis of TTP and leading to discontinuation of plasmapheresis and initiation of a therapeutic trial of eculizumab for presumed aHUS, pending complement genetic testing. Eculizumab leads to prompt improvement in the patient’s hemolysis and renal function. Subsequent genetic testing revealed heterozygosity for a c.481C > T mutation in the C3 gene (p. Arg161Trp), as well as the disease-associated factor H CFH–H3 and membrane-cofactor protein (MCP) haplotypes. The patient has tolerated eculizumab treatment well with improvement in his laboratory parameters (Table 1).
Patient 2 is a 34-year-old Caucasian woman with an unremarkable past medical history who presented to the clinic with vaginal bleeding. Six days prior to presentation, she started experiencing nausea, vomiting and diarrhea, for which she went to a local clinic and received symptomatic treatment. The diarrhea soon began to improve, when she started noticing unusual mid-cyclic vaginal bleeding that was significant. On initial assessment, her blood pressure was 204/110 and she was afebrile, with otherwise unremarkable vital signs. She had blurry vision in her left eye, but no other focal neurological deficits. She was transferred to the emergency department for further evaluation. On examination, the patient was alert and oriented. She appeared mildly distressed. She was notably hypertensive, with no other signs of end organ damage. The initial work-up was remarkable for normocytic anemia, thrombocytopenia and elevated serum creatinine, with otherwise stable serum electrolytes, liver enzymes and bilirubin levels (Table 1).
The treatment for hypertensive emergency was started and she was admitted to the intensive care unit for further care. Further work-up results raised concern for intravascular hemolysis, given anemia, thrombocytopenia, elevated LDH and decreased haptoglobin, but the peripheral smear revealed few schistocytes. Given the overall presentation, an evaluation for thrombotic thrombocytopenic purpura (TTP) was undertaken. The ADAMTS13 level was > 100% and complement C3, C4 and CH50 levels were normal. A renal biopsy was performed and the histopathology showed evidence of significant thrombotic microangiopathy. An aHUS complement panel revealed sC5b-9 complement of 279 (Reference: <251 ng/mL), and CBb complement of 3.7 (Reference: <1.7 mcg/mL), which was consistent with recent activation of the alternative complement pathway. The patient was started on weekly eculizumab for possible aHUS, along with hemodialysis and aggressive blood pressure control. Genetic testing subsequently revealed a heterozygous variant in the C3 gene (c.481C > T). The eculizumab was continued and her renal function slowly improved, ultimately leading to the discontinuation of hemodialysis.
DiscussionIn contrast to the TTP and typical HUS, complement-mediated aHUS can be caused by a variety of triggers and is often precipitated in patients with congenital or acquired defects in regulatory proteins in the alternative complement pathway. It is estimated that approximately 50% of patients with aHUS have underlying mutations in the genes encoding complement proteins.3
The complement system is an essential part of innate immunity, designed to fight infections and to handle damaged cells and debris. It is characterized by a fine balance between the continuous complement activity, required for fighting pathogens, and complement inhibition to protect the host from excessive damage.3 Dysregulation of this balance can result in an inadvertent host damage. This dysregulated complement activation can trigger an inflammatory cascade at the endothelial surface with subsequent microvascular endothelial injury. Microthrombi can eventually form, causing red blood cells to sustain high levels of shear stress, as they flow through those turbulent areas with endothelial injury. The affected microcirculations ultimately get partially occluded, causing ischemic end organ damage.
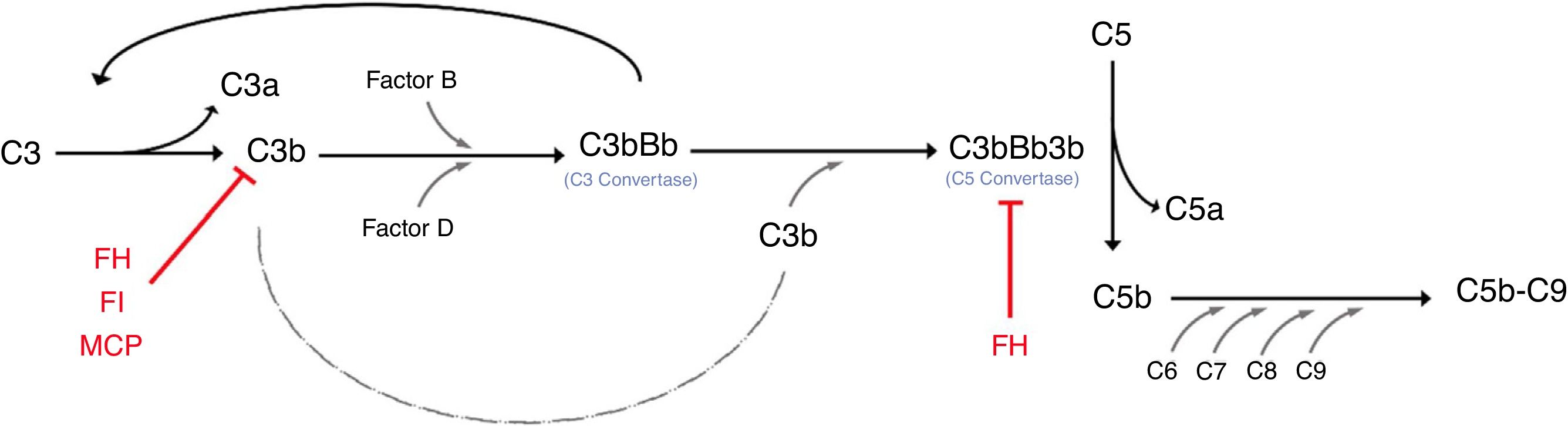
Activation of the complement system occurs via different pathways, ultimately resulting in the enzymatic cleavage of the central component C3 through C3 convertase. The C3 convertase of the alternative pathway, C3bBb, is composed of the active fragments of C3 and factor B (FB). To prevent uncontrolled activation and subsequent host tissue injury, the complement system is tightly controlled by surface regulators, such as factor H (FH), factor I (FI) and membrane cofactor protein (MCP; CD46) (Figure 1). In aHUS, these regulators are often mutated and unable to efficiently protect the endothelium from complement activation. In the presence of events that enhance alternative-pathway activation, carriers of complement mutations undergo excess C3b formation and deposition on the vascular endothelium. An uncontrolled level of C3 convertase leads to more C3b molecules and to more C5 convertase, initiating the formation of the membrane-attack complex (C5b-9). The complement-mediated endothelial injury creates a prothrombotic state, through exposure of subendothelial collagen, von Willebrand factor and fibrinogen.3
. The cleavage of C3 allows Factor B to bind to C3b and subsequently get cleaved by Factor D. The result of this series of reactions is the formation of C3bCBb, which is known as C3 convertase. The C3 convertase can form an amplification loop, resulting in an exponential cleavage of C3. The binding of the C3b molecule to the C3 convertase produces C3bBb3b, which is C5 convertase). The C5 convertase then cleaves C5 into C5a and C5b. The C5b joins C6, C7, C8 and C9 and assembles the membrane attack complex (C5b-C9). The regulator of the alternative complement pathway (red) includes Factor H (FH), factor I (FI) and the membrane cofactor protein (MCF).3")
Depicting the alternative complement pathway and regulators of excessive activation.
The alterative pathway is constitutively active because of the spontaneous cleavage of C3 (a process referred to as tickover). The cleavage of C3 allows Factor B to bind to C3b and subsequently get cleaved by Factor D. The result of this series of reactions is the formation of C3bCBb, which is known as C3 convertase. The C3 convertase can form an amplification loop, resulting in an exponential cleavage of C3. The binding of the C3b molecule to the C3 convertase produces C3bBb3b, which is C5 convertase). The C5 convertase then cleaves C5 into C5a and C5b. The C5b joins C6, C7, C8 and C9 and assembles the membrane attack complex (C5b-C9). The regulator of the alternative complement pathway (red) includes Factor H (FH), factor I (FI) and the membrane cofactor protein (MCF).3
In both cases we present here, the patients were carrying the same C3 heterozygous missense mutation, c.481C > T (p.Arg161Trp). Functional studies demonstrate that this variant results in a hyperactive C3 convertase, with an increased affinity for factor B, along with a decreased binding to the MCP.4,5 One study has found a higher incidence of cardiac (60%) and neurologic (35%) events in patients with aHUS with the underlying C3 mutation, findings that were not evident in our patients. However, the exact clinical implications of this mutation on the disease behavior and prognosis have not been very well characterized, primarily due to the paucity of data in the literature.4
This specific gain-of-function mutation in C3 has been reported previously at a frequency of 1–4% in individuals with aHUS and in up to half of the C3 mutations associated with aHUS in Europe.4 Furthermore, most of the carriers of this variant worldwide were found to be of European descent. Given its relatively high frequency in certain populations, this specific mutation could be considered a prototypical aHUS C3 mutation.4–6
In contrast to HUS and TTP, plasmapheresis in aHUS has been associated with variable efficacy with a high rate of progression to end-stage renal disease and graft complications after renal transplantation.7 Eculizumab, a recombinant humanized monoclonal antibody directed against C5, has successfully induced remission of aHUS in multiple case reports and is frequently utilized in aHUS.8
The risk of relapse after eculizumab discontinuation is high with unpredictable timing. Available data suggests indefinite therapy remains the safest recommendation. Prospective data is lacking to adequately stratify risk of relapse after eculizumab discontinuation. Early observational studies have shown that risk may be increased in certain complement protein mutations. One study has showed that increase in the time interval between eculizumab infusions, guided by complement activity monitoring, did not change the disease activity.9 More studies are needed to further characterize predictors of relapse and help develop validated tools to guide dosing and course of eculizumab treatment in aHUS patients. This may allow a safe reduction in the frequency of eculizumab administration, as well as help reduce the high cost associated with eculizumab therapy.
Many other treatments that target the complement cascade are currently undergoing clinical trials. For instance, in a phase 3, open-label study, published by Lee et al. in 2018, ravulizumab given every 8 weeks has shown noninferiority, compared to eculizumab in complement inhibitor–naive adults with paroxysmal nocturnal hemoglobinuria (PNH), with a similar safety profile10; this agent represents another therapeutic option for patients with aHUS.
ConclusionIn conclusion, we report 2 cases of aHUS most likely driven by the same gain-of-function mutation of the C3 protein. These cases and the existing literature support the importance of recognizing this pathological variant as a cause for aHUS. Both patients responded well to the eculizumab therapy and have been in remission, with a gradual decrease in the frequency of eculizumab administration. Further studies on such patients might characterize disease behavior and prognosis specifically related to this mutation.
FundingThere is no funding support for the conduct of this article.
Conflicts of interestThe authors declare no conflicts of interest.


