



Advanced therapy medicinal products, considered special medications, requires Anvisa approval for use and commercialization in Brazil. They include the advanced cellular therapy products, tissue engineering products and gene therapy products, which due to their complexity involve innovation and risks, optimized regulatory channels for their development and life cycle monitoring. The scientific elements and the compliance with applicable regulatory aspects are fundamental pillars for the advancement of clinical trials, the positive evidence of the benefit-risk profile and the definition of the critical quality attributes, from the perspective of making safe, effective and high-quality products available to the population. The approval models of these products in Brazil adapt to the specificities and characteristics of the technology and the patient target population, with accelerated regulatory analyses, use in emergency situations by risk controls and specific monitoring mechanisms, principally those related to rare diseases without other therapeutic alternatives. The opportune access to the advance therapy product with safety, efficacy and quality involves innovative normative elements that include the long-term follow-up of the safety and efficacy and of the adaptive pharmacovigilance requisites, as well as the traceability mechanisms for the start-off materials, products and patients.
Blood, tissues, cells and human organs constitute themselves on therapeutic alternatives, regulated in Brazil as products of close vigilance, converging in a regulatory model which is internationally prevalent.1–4 Not being eligible for marketing authorization at Anvisa, produced or manipulated in differentiated health establishments, the sanitary control of quality and safety of these products is derived from the inspection actions of Good Practices applied to each productive, and therapeutic use chains.5-7 On the other hand, technically obtained or elaborated pharmaceutical products which have prophylactic, curative and palliative finalities, denominated medications, are registered in Brazil and in the world by means of exhaustive proof of their safety, efficacy and quality with a national competent authorities.8,9 Contrarily, as for the similarity in therapeutic functions of products based on blood, tissues, cells and organs and the conventional medicines, the regulatory models of the two are differentiated according to the risk potential, technological development and innovative clinical use.
The advances in biotechnology led to a new perspective on innovative products that use the cellular and genetic potential, by means of cellular culture techniques, materials sciences and recombinant DNA technology.10,11 In this context, a class of products based on human cells, tissues and genes has been developed and approved as a new therapeutic arsenal.12 It is denominated Advanced Therapy Medicinal Products (ATMPs) destined to the treatment, prevention or diagnostic support for diseases, including advanced cellular therapy products, gene therapy products and tissue engineering products, which may be combined or not with medical instruments.13-17
A fragilized regulatory environment places at risk the health of people by means of permissiveness in the indiscriminate use of unsafe products without proven efficacy, aggravated by profound asymmetries in information and negative externalities. Added to this scenario is the dangerous direct marketing to the consumer on the interventions with stem cells and other non-approved advanced products.18-21 According to available reports in the EuroStemCell,18 in Germany prior to 2012, diverse companies and medical centers were observed promising a cure for untreatable diseases, such as Parkinson's, Alzheimer's and others, with autologous stem cell injections, alleging exaggeratedly promising effects, with high cost and sanitary risk.19-21
According to the Pan-American Health Organization (PAHO/WHO),15 in the Americas region there is a similar phenomenon of proliferation of medical advertisements that promise the population access to cell therapies, which in many cases does not have the minimal scientific substantiation in safety, efficacy and quality, entailing critical clinical risks to the patients. In this sense, the Food and Drug Administration (FDA), the American regulatory agency, under alert against the indiscriminate use of cell therapy in the United States,22 has informed that unproven and unauthorised stem cell therapies can be particularly unsafe for patients. In an article in The New England Journal of Medicine (2017),23 on the benefits and risks of cellular therapy, the FDA states that clinical trials with these products are principally derived from small uncontrolled studies containing hardly reliable reports on the efficacy. According to the Agency, the scientific literature is full of clinical case descriptions of therapeutic interventions with cells that, in ultimate analysis demonstrated their ineffectiveness or harmfulness, when scrutinized in well-controlled trials. Likewise, the European Medicine Agency (EMA),24 in a document published in 2020, states that “patients who use unproven or unregulated therapies based on cells and genes, have suffered grave collateral, sometimes fatal, effects, including infections, undesired immunological reactions, tumor formation (…)”, highlighting that cells infused in patients with the objective of exerting a function distinct from the original or cells which have been substantially manipulated aggregate risks and should be regulated with the same regulatory vigor as medications.
Concerned about the internationally reported situation and detecting similar cases in investigative processes in Brazil in 2012, Anvisa initiated an ample scientific and social discussion, also of a judicial nature,25 on the advanced cellular therapy regulation, which would culminated in 2018, with the publication of the first Brazilian sanitary norms applied to the ATMP, determining for these products the concept of “special medications”. Considering the international regulatory convergence, Brazil has been structuring the infraconstitutional normative benchmark with the establishment of rules and assumptions for the manipulation of cells and human genes and their transformation into products with therapeutic finality, to be registered and commercialized, intending to dignify the life of those who might be treated with them.17 Highlighted is another crucial point derived from Brazilian constitutional precepts, the guarantee of gratuitous collection of cells, tissues and other body parts to be employed as start-off materials in the manufacture of the ATMP, by free, spontaneous and informed donation, so as to dispel the risk of any abuse.17
The objective of this article is to briefly present the Brazilian regulatory model applied to the ATMPs, considering their development cycle, approval and post-commercialization monitoring. This is an exploratory study of documental revision. In addition to the related scientific bibliography, official documents of Anvisa, FDA, EMA and others pertinent to the ATMP regulation were also referred to.
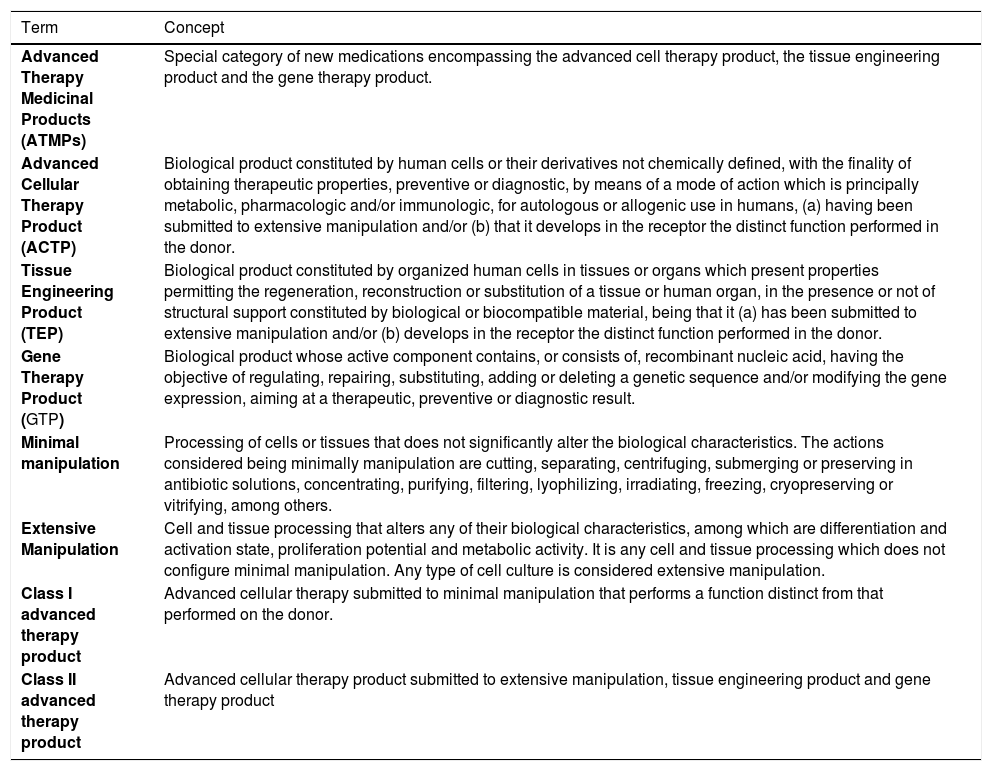
Regulatory conceptsGoods and products subject to the sanitary vigilance are those which involve the possibility of risk to the public health, including those obtained by means of genetic engineering or other biotechnological procedure.26 It is in this legal context, determined by Law 9782/99,26 that the ATMPs are included for clinical trials or therapeutic use in Brazil. It was in the same vein followed by the EMA13 and FDA14 that Anvisa16 in Brazil defined the advanced therapies products as a special category of medications, subject to the same regulatory mechanisms.27,28 The definition of the ATMP in the Brazilian norms is fundamental to establish the requisites for the mitigation of sanitary risks. The products categorized for regulatory ends and their concepts are described in Table 1.
Regulatory definition for advanced therapy products in Brazil, 2021.
Source: Anvisa Resolution 505/2021.29
Using these regulatory concepts establishes a differentiation between products based on cells and tissues considered advance therapies and products which involve cells, tissues and human organs, as in conventional therapies applied in transfusion, transplant and grafting procedures. The core of the differentiation of therapeutic products based on human body parts is determined by the technological increment and innovative clinical indication present in the ATMP. For example, the selection of cells by peripheral blood apheresis to obtain a greater concentration of hematopoietic progenitor cells (HPCs) for transplant is considered a cellular therapy with minimal manipulation, these cells being used in the same essential function and the same anatomy or histology of origin. Using the same example as the HPCs, when these cells undergo multiplication and differentiation in culture, under specific conditions, it is considered a substantial or extensive manipulation because the characteristics of cellular multipotency and capacity for self-renovation are altered.30
An important derivation from the concept of gene therapy product (GTP) is the attribution of its therapeutic, prophylactic or diagnostic effect directly on the recombinant nucleic acid sequence or on the genetic expression product of this therapeutic sequence. These types of ATMPs encompass the products that impart their therapeutic effects by transcription and/ or translation of genetic material transferred by means of transfer vectors (viral or nonviral) to the cells of the patient, in the in vivo or ex vivo modalities. It must be highlighted, however, that the vaccines against infectious diseases based on the genetic material of microorganisms are not included in the in the GTP class.31 Another example that does not qualify as GTPs are the products based on wild oncolytic viruses if these do not possess recombinant nucleic acid.32 Furthermore, it is also important to highlight the regulatory point of view that genetically modified T-cell-based products, the chimeric antigen receptor T-cells (CAR-T), are classified as ex vivo gene therapy products.
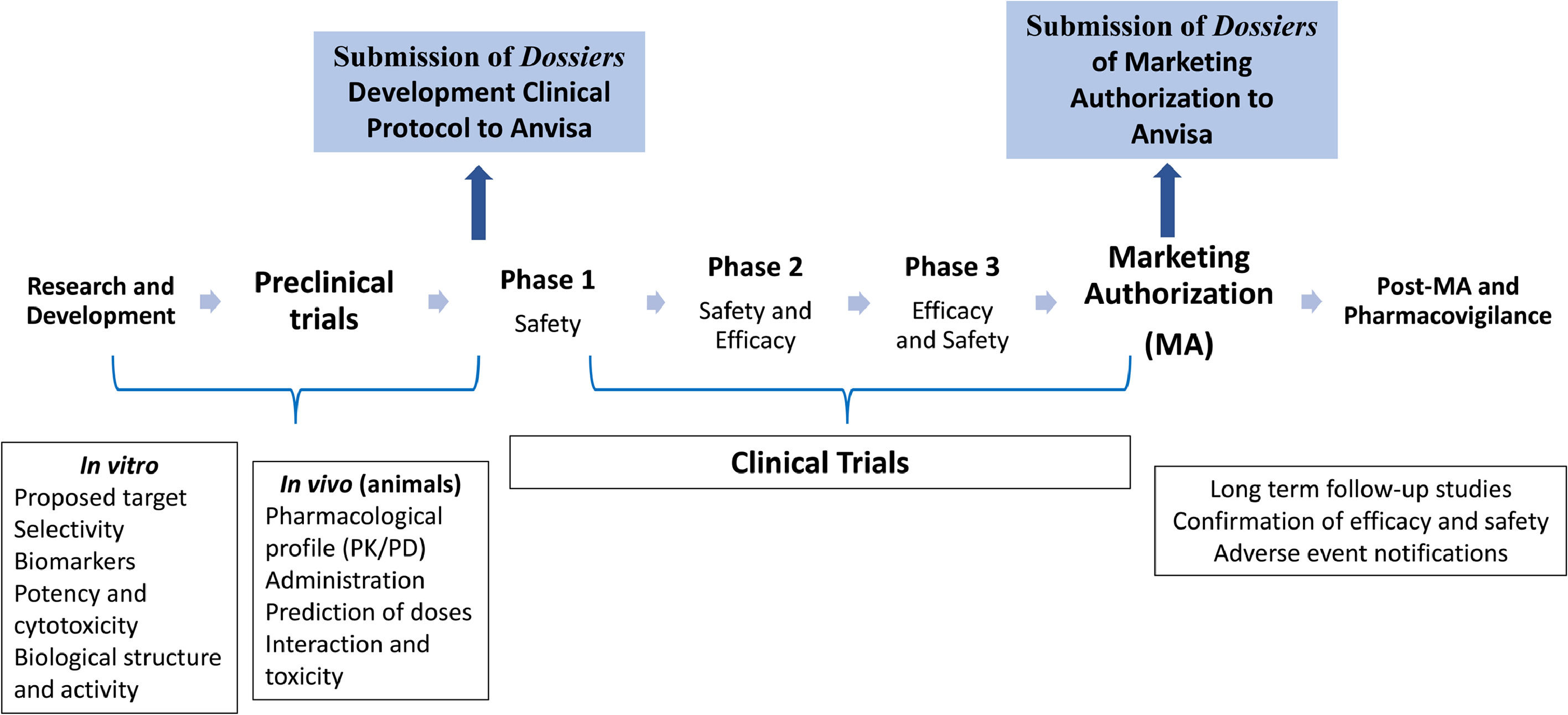
Regulating advanced therapies in BrazilAs determined by Brazilian sanitary laws, no ATMP, including the imported ones, can be commercialized or administered to the patient prior to registration with Anvisa.33 The ATMP marketing authorization in Brazil follows the same structure and approach used for any other medication, as schematized in Figure 1. The interested company must demonstrate: (1) the quality aspects (characterization, production and control process and risk management; (2) the security profile (pharmacological/toxicological characteristics) and proof of product concept in vitro and in vivo models, and (3) the safety profile in humans and the results of efficacy for the indication(s), dosage(s) and established target population(s).29

Anvisa initiates the regulatory follow-up process of the development of a new product in the clinical trials phase. Always, when there is the intention to initiate clinical studies with a new ATMP in Brazil, it is necessary to obtain previous approval at Anvisa, even when a new indication or different administrative path for a previously registered product is being studied. The Agency concedes the permission to perform clinical research on the investigational product with the objective of determining the elements for its clinical safety, efficacy and quality attributes, under a process conducted in a controlled form and by responsible sponsors and researchers.34
The scientific elements and the compliance with regulatory aspects are fundamental pillars for the advance of the developmental processes of a therapeutic product. In Brazil, three public institutions act with complementary functions in the supervision of clinical trials on an ATMP. The approval of ethical and social aspects of studies involving human beings is of the responsibility of the National Commission on Ethics in Research (in portuguese, CONEP), by means of the CONEP System. In parallel, Anvisa evaluates the quality and safety aspects of the investigational product by means of a criterious revision of fabrication (and its controls), the characterization tests (identity, purity, potency and others) and the sterility of the finished product and intermediaries of the process, as well as the scientific validity of the proposed protocol, or in other words, the capacity of the designs of proving the efficacy and safety of the product, so as to permit an evaluation of risks and benefits. It must be highlighted that, in the clinical development phase of gene therapy products, it is important to evaluate the biosafety of the component identified as a genetically modified organism, performed by the National Technical Commission on Biosafety (in portuguese, CTNBio).34
A clinical trial dossier should be composed of the following documents: 1) clinical investigation plan for the investigational ATMP which contemplates information on the clinical development process of the product, 2) specific protocol of the clinical trial to be performed in Brazil, 3) investigator's brochure containing the minimum information on the pre-clinical and clinical data of the product and 4) the documents proving the production process quality. At Anvisa, particularly in the Blood, Tissues, Cells and Organs Management (in portugueses, GSTCO), the ATMP dossiers are evaluated by teams of specialists, who may at any moment request the ad hoc evaluation of external specialists registered in the Anvisa Network of Specialists in Advanced Therapy (in portuguese, RENETA), or members of the Committee for Advanced Therapy (CAT), depending on the product and its clinical application. The general flow of document submission to Anvisa forecasts analysis deadlines of the Agency by type and technical complexity of the dossier, being 180 days for the Clinical Advanced Therapy Product Development Dossier (in portuguese, DDCTA) and 30 days for the Simplified Clinical Advanced Therapy Product Development Dossier (in portuguese, DSCTA) . The definition of deadlines for the conformation of the evaluative and decisional process at Anvisa and the responses and adaptations of the sponsor is an important factor for the transparent and predictable regulatory process.35
For the initiation of the clinical trials, it is essential that robust and specific pre-clinical research data, preferentially performed in a setting of Good Laboratory Practices (GLP) with the investigational product, be provided. Aiming at regulatory analysis, it is important for the sponsor to demonstrate how the pre-clinical studies and respective results were performed, including the pharmacology and toxicology studies, be they in vitro or in vivo, preferentially in relevant animal models. Such studies should generate data permitting the verification if the product is reasonably safe for initial tests on humans. The pre-clinical data should be adequate in their support of the proposed clinical trial with recommendations for the safe dose, scaling scheme and administration route.36 Previous experiences of the product in patients, when existent, even in emergency use situations, can be incorporated to the information to corroborate the pre-clinical data. In terms of genetic therapies, specific questions should be studied in the pre-clinical phase, for example, concept proof studies, product biodistribution, level and persistence of genetic expression, alteration in germinative lineage and others. The toxicology studies should also take into consideration the risk of vectors used and their potential for insertional mutagenesis.
The basic structuring of clinical development is generally divided into three phases, with each supplying subsidies and support for the next stage, with robust data which corroborates the positive balance between benefits and risks.36 One of the phases of important attention to the regulator is when the ATMP is used for the first time in human beings, which denotes an element of higher risks due to the uncertainties, unpredictabilities and limitations of the pre-clinical data in the prediction of safety and efficacy. The principal focus in a phase I study is the monitoring of the product safety in a specific population of few patients, though it must be noted that the safety evaluation of a product remains being investigated during the whole development. These studies should be projected to determine the metabolic and pharmacologic actions in humans, obtaining sufficient information on the pharmacokinetics, associated adverse events and dosage schemes.36
The innovative characteristics of the ATMPs related to pharmaceutical biotechnology and the indications for rare conditions without therapeutic alternatives has challenged the researchers in the proposition of trial designs with combined phases, for example, phase I/II, phase II/III and additional trials in phase III following the register. For many ATMPs, the phase I trials are conducted objectivizing the evaluation of safety (primary objective of this type of study), frequently combined with an early efficacy evaluation (considered in the secondary objectives); another possible design combines the dose scaling phase with the initial efficacy evaluation, by means of the cohort expansion of the patients who received the dose considered safe in the scaling stage, permitting that efficacy data on patients in phase I be grouped with the efficacy data of patients in phase II, resulting, therefore, in a phase I/II study design. The small populations of patients also permit that the researchers experiment with innovative trial projects, with the involvement of Anvisa from the initiation of the process, including endpoints, new or substitutive, and single-arm trials, with the use of the natural history of the disease data as the comparative group. Randomized clinical trials are amply accepted to supply more reliable evidence upon evaluating the safety and efficacy of a new intervention and obtaining regulatory approvals.36-38 Nevertheless, it will not always be possible to rigorously follow the randomized clinical trials rules, principally for rare diseases that have few to no effective treatment options available, making important the consideration of other measures which can aid in improving the strength of scientific evidence applied to the ATMPs. Under discussion is the use of literature data to support safety prospection decisions and concerns, in addition to the natural history studies which assist in the outcome evaluation of efficacy proposals. In the particular case of advanced therapies, the information on the fabrication process and the release criteria are also necessary to adequately correlate the literature results used with the exclusive product characteristics.38
Valid scientific principles should be applied in all ATMP development phases in relation to safety, active components, raw materials, starting materials and fabrication process quality control. In the case of ex vivo gene therapy products, for example, CAR-T cells, a dossier should be drawn up for Anvisa evaluation, with the description of the cell source, clinical and laboratory screening of donors, collection and processing methods, culture conditions and procedure for the genetic cell modification procedure. Extensive safety trials and modified cell characterization must include information on the cellular identity and viability, cell subpopulation percentages, transduction efficacy, genetic expression longevity, transgene integrity, genetic stability during the in vitro or differentiated proliferation, number of copies of the transgene per transduced cell and presence of competent replication virus, as well as the sterility, mycoplasma and endotoxin evaluations.39 Care must also be taken when dealing with autologous products so that adequate labeling and tracking systems can be used. An initial stage in the development of a GTP is the construction of the vector responsible for the delivery of gene of interest to the target cell. This vector can be tested and rigorously characterized. The information supplied in the regulatory documentation on the vectorial construction should include the description of the vector components, their source and derivation, restriction and cloning site, regulatory elements and others, as well as their production and purification, identity, quality, purity and potency.39 From the initial developmental phases onwards, the analytical trials qualification should be established to permit the collection of reliable data and the development of acceptance criteria which are adequate for the quality control of raw materials and starting materials.
Another fundamental factor in the process of clinical trials information on the qualifications of the researchers that supervise the administration of the experimental product and detect and manage the adverse events. Moreover, attention must be given to the standardization of the product preparation processes prior to its administration and to the training of all professionals involved.34
When Anvisa receives insufficient or incomprehensible information in the documentation which impede the understanding and the adequate evaluation of risks and potential benefits, the appropriate regulatory demands are made. For the pursuance of the regulatory analysis, the sponsor must correct the identified deficiencies or provide supplementary information. Critical situations can lead to the non-approval of the clinical trial, for example, if it is detected that patients or participants in the research will be exposed to risks and irrational and significant harm.
Following the consent by Anvisa, attending to the other Brazilian legal requirements, the clinical trial can be initiated, following the Good Clinical Practices (GCP) rules. Annually, the sponsor must forward monitoring reports to the Agency, including adverse events experiences and partial summaries of the results obtained. It is furthermore highlighted that grave adverse events which are life-threatening and deaths must be opportunely reported to the Agency, the maximum deadline being 7 (seven) days from the date of awareness of the case, the remaining grave adverse events being reported within 15 (fifteen) days.34 During the evaluative process by Anvisa on certain ATMPs, a case-by-case approach is applied, to guarantee that national requisites and international recommendations be attended to in an appropriate manner.
An investigational ATMP can be used out of a clinical trial before its registry approval by means of specific programs, such as the expanded access (group of patients), compassionate use (specific patient) and post-study supply, with the previous approval and supervision by Anvisa.40Table 2 below presents the concepts applied to the access to investigational ATMPs, as per the Anvisa resolution 38/2013.
Principal concepts of access programs to investigational products in Brazil, 2021.
Source: Anvisa resolution 38/2013.40
An ATMP can only be authorized if the benefit-risk profile is positive, the benefits being related to the principal favorable effects of primary and secondary clinical outcomes, while the risks describe incidence, gravity, duration, reversibility and dose-response relation of unfavorable effects and adverse events of the product. Clinical study limitations related to the size of the sample and representativity of the patient target population must be adequately discussed and will be taken into consideration in the decision-making.29
The demonstration of safety and quality of the product requires the implementation of specific tests, pharmacopeial or validated tests, which evaluate identity, potency, sterility, purity, qualification and quantification of impurities, presence of mycoplasmas, endotoxins, adventitious viruses and others.41,42 The product identity is demonstrated through specific identification trials and its distinction from any other substance. In turn, the potency tests should indicate the specific capacity of the product of reaching a determined result. Another important aspect, the ATMP purity, can be defined as the capacity of detecting foreign elements or materials present following the fabrication processes. The purity test of a gene therapy product, for example, involves trials for the detection of viral capsid residual proteins, host cell residual DNA, RNAs, competent replication virus, undesirable nucleotide sequences, solvents, cryopreservatives or auxiliary products of production and purification, such as cytokines, antibodies, antibiotic residues, culture media, serums and others. To complete the characterization of the product, a dossier should be presented on the stability tests program, in accordance with each type of product, for example, the product of advanced cellular therapy and ex vivo GTP, including the counting and cellular viability, sterility and potency tests.41,42
Other requisites which should be completely defined are critical quality attributes and the Good Manufacturing Practices (GMP) Certification issued by Anvisa for the fabrication of the active component and final ATMP. Principal GMP elements include the maintenance of detailed registers, standardized written procedures (POPs), quality and analytical trials control program, qualification of suppliers and equipment, process validation, personnel qualification and training program, certification of installations and environmental monitoring.43,44 The stability studies on intermediary and finished products should be appropriate for the definition of the validity deadline and proposed filling system.43,44 Total adhesion to GMPs, required in the marketing authorization process, provide quality and safety in the process and permit a reproducible and consistent performance of commercial product lots.
A manufacturer must describe any change in the product, independent of occurring prior to, or following, the registration. The change in the fabrication process must be evaluated and the resultant product compared to the existing product to guaranteed that the change has not altered the safety, purity, potency or integrity of the therapeutic product or any other quality characteristics compromising its clinical performance. The comparability studies can be based on a combination of in vitro or in vivo studies, pharmacokinetics or pharmacodynamics evaluation, toxicity in animals, clinical tests and others, depending on the alteration reach. The product comparability must be demonstrated by means of comparative analyses of lots of products manufactured according to the prior and new procedures. In these cases, Anvisa evaluates and determines, in light of the results presented, if the comparability data are sufficient or if additional studies will be needed. Examples of changes requiring a comparability study include fabrication site alteration or critical alterations in productive flows, changes in the cell or virus banks, vector modification, cellular culture alterations, isolation or purification and changes in the storage recipient or product formulation, among others.44 In Table 3, there is a brief presentation of the essential documents that must be presented to Anvisa for ATMP marketing authorization.
Principal documents in the ATMP registration dossier in Brazil and examples of information to be discussed. Brazil, 2021.
Source: Anvisa Resolution 505/2021.29
A approval for the standard ATMP marketing authorization is conceded when the information on quality is adequate and the data proving the efficacy and safety are unequivocal, statistically significant and based on clinical trials which are complete. Exceptionally, there is the ATMP approval conceded under certain conditions which requires further confirmation of long-term clinical efficacy.29 For this conditional approval, the ATMP must obligatorily attend to a clinical necessity without treatment or clearly offer a therapeutic advantage over available treatments. Additionally, it should be analyzed if the product proposes to attend to a grave or rare and debilitating condition or in situations of risk of death or yet, in public health emergencies. The exception occurs when the benefit of immediate availability supersedes the product risks and the fact that additional evidential data on its long-term clinical efficacy are still needed.29 Evidence obtained by means of a substitute clinical outcome, such as a biomarker, rather than a direct therapeutic measurement, can be accepted for the register under certain conditions, as long as the substitute outcome be adequately validated, In some cases, additional trials will be necessary, with real clinical outcomes, that can be ongoing or forecast to be performed, whose results may be presented on defined dates in terms of commitments established between the bearer of the register and the Agency. It is also necessary to evaluate if the register bearer has the conditions and planning to supply data and information annually to Anvisa, during the time established for monitoring. For example, the two gene therapy products registered at Anvisa in the year 2020 (Luxturna® and Zolgensma®),45 for the treatment of rare diseases, were conceded under conditions with obligations and commitment terms that will extend into 2038, requiring annual renovations until then.
The Brazilian normative29 defined the possible use of an ATMP not liable to the register in an emergency situation under medical responsibility, with a specific prescription for a determined patient. This regulatory recourse was developed in Brazil for exceptional situations in which a determined product, not being under clinical development, may attend as an emergency a patient in the condition of risk of death in treatment for diseases without therapeutic alternatives available in the Country. This type of experimental product cannot be commercialized, its clinical use based on the scientific rationale and previous clinical experience of the doctor. In addition to the individual control performed by the responsible professional, it is fundamental that the product be produced under the GMP principles in all fabrication operations of the active component and de final product. For example, the use of experimental ex vivo GTP must be previously authorized by Anvisa with the submission of summarized technical information, in addition to the Free and Informed Consent Term (FICT) signed by the patient or legal guardian.29
In the Brazilian legislation, the category of priority product was established, with an accelerated rite of evaluation that encompasses ATMPs destined to the treatment of rare and neglected diseases, for serious and debilitating conditions in which there is no available therapeutic alternative and for public health emergencies. In this category, the Anvisa evaluation period was reduced from 365 to 120 days. The central idea of the regulatory acceleration process was to guarantee that patients in specific situations have the opportunity to benefit from new treatments. Another type of ATMP that can be included in this priority category is that which is destined to offer a new therapeutic indication to the pediatric population. Also having analysis priority are the ATMPs which had clinical trials (phases I and II) developed in Brazil. In all situations, the solicitation for accelerated analysis should present in their dossiers adequate justifications for their inclusion in these categories. Additionally, it is recommendable that an initial pre-submission dialog between the company and the Agency already have been initiated.
It is important to highlight that the accelerated process or one under conditions have the same quality requirements as a standard marketing authorization, being necessary to demonstrate that the fabrication process is robust, reproducible, validated and by means of a GMP Certification.
Despite the type of register conceded and the agreed upon monitoring conditions, any significant alteration of the data submitted to Anvisa should be notified and on determined occasions be previously approved by the Agency prior to their implementation. This update with the Agency is of fundamental importance to permit dynamicity in the product's life cycle while ensuring regulatory control.
Following the ATMP marketing authorization approval, studies may be necessary to supply continuous evidence of the positive risk-benefit equilibrium, along with data on the real use in patients. The post-register studies are continual product development strategies, which include clinical studies for a new indication, amplification of the target population, comparability of the product following fabrication changes, validation of substitute clinical outcomes, demonstration of the superiority of other treatments, etc.
General aspects of pharmacovigilancePharmacovigilance is related to the identification, evaluation, comprehension and prevention of adverse effects or any problems related to the use of medications commercialized in the Brazilian market, including adverse events stemming from quality deviations, therapeutic ineffectiveness, medication errors, use of medications for indications not approved in the register, abusive use, intoxications and medication interactions, among others.46 Pharmacovigilance processes should be applied to the ATMPs approved with Anvisa, as well as with post-use monitoring mechanisms. The Risk Management Plan (RMP) is an indispensable requisite in the dossier,29 which should address elements involving the safety profile specification and identification of potential risks to be managed or studied in the post-register. The RMP for the ATMP should also address the specific risks associated, for example, with donors of starting material, with surgical or administration procedures, with the risks involved in the transmission of germinative line vectors and others.47
PerspectivesAnvisa has the fundamental mission of promoting efficient regulation to attend to the innovations involved in the Advanced Therapy Medicinal Products, with the objective of permitting the patients access to the new effective, safe and high-quality products. It is also in the Agency's role to supply necessary information to patients and healthcare professionals with transparency and clarity, based on regulatory science and on approved products information, thus decreasing the asymmetries. The regulatory evaluation is based on robust scientific substantiation and on constant analysis of the risk-benefit profile, ensuring the safety of the patients, but also permitting the development of innovative products. Strategies to improve the comprehension of the regulatory aspects of advanced therapy products forecast the participation by Anvisa in scientific forums, sector discussions and dialogues with researchers and patients, with the perspective of developing the best regulatory practices and providing the development of safe, quality and effective innovative products with focused on the positive impact on the life of Brazilian patients.


