

Currently, there are four CAR-T products commercially available on the market. CAR-T cells have shown high remission rates and they represent an effective treatment option for patients with resistant or refractory B cell malignancies. Approval of these cell therapy products came after an extended period of preclinical evaluation that demonstrated unprecedented efficacy in this difficult-to-treat patient population. This review article outlines the main preclinical evaluations needed for CAR T cell product development.
T-cell expressing chimeric antigen receptor (CAR T-cell) has emerged as promising therapy for hematological malignancies. Since 2017, four CAR T-cell products - axicabtagene ciloleucel (axi-cel), tisagenlecleucel (tisa-cel), lisocabtagene maraleucel (liso-cel) and brexucabtagene autoleucel - have received the approvement by US Food and Drug Administration (FDA) for treatment of relapsed/refractory (R/R) Non-Hodgkin B-cell lymphoma (B-cell NHL). At the same time, tisa-cel has been approved by the FDA for R/R acute lymphoblastic leukemia (ALL) treatment.1
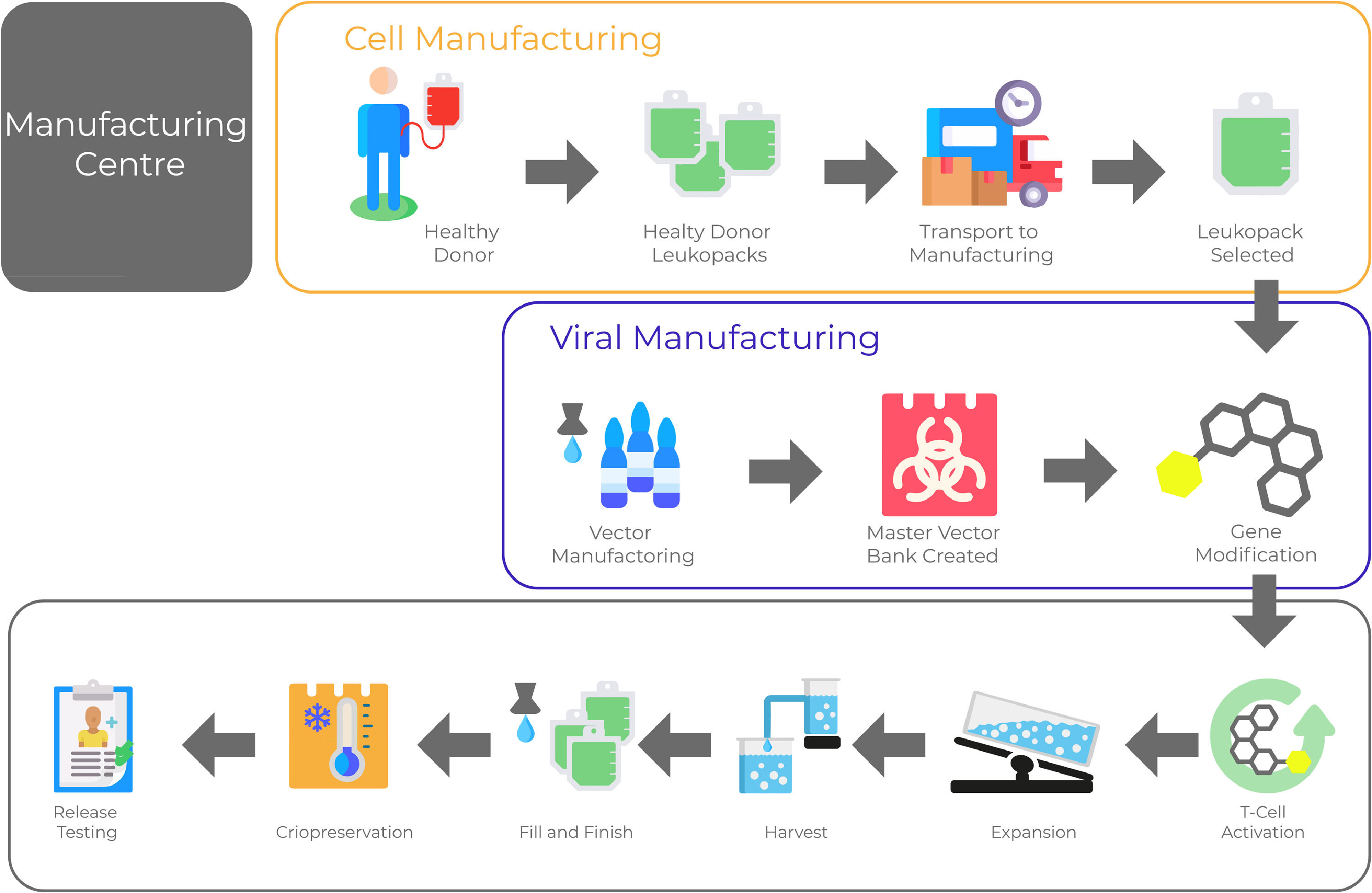
One crucial aspect of CAR T-cell therapy is to establish and validate a reproducible manufacturing process of high-quality able to deliver clinical-grade CAR-T cell products. It is a prerequisite for the wide application of this technology. Product quality needs to be built-in within every step of the manufacturing process (Figure 1).

However, before the clinical-grade platform validation, many preclinical assays have to be performed. There are several imperative experiments requested by different regulatory international agencies - for instance, FDA and European Medicines Agency (EMA) - as essential pre-step data to establish a robustic investigational new drug (IND) dossier. In Brazil, the Agência de Vigilância Sanitária (ANVISA) incorporated on its regulatory framework the same principles of FDA and EMA.2
There are great expectations for the imminent introduction of CAR-T therapy in Brazil. Its significant complexity, however, represents an important challenge for proper implementation and achievement of the same goals observed in other countries. This consensus is based on the expert opinion that appointed the minimum preclinical assays required to support the development of new CAR-T cell therapies.
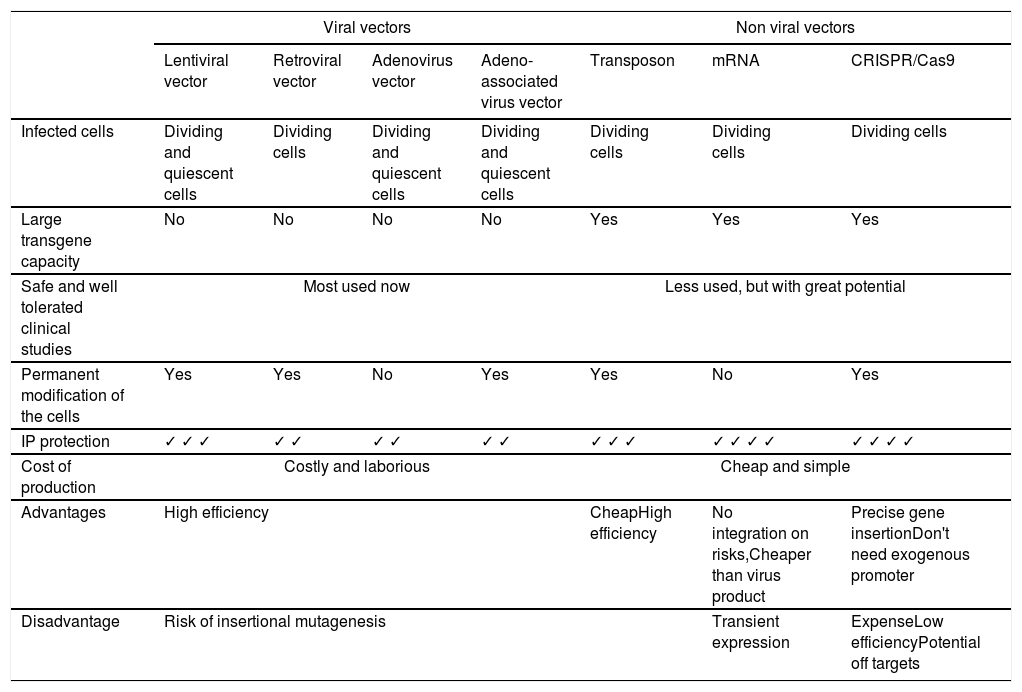
Vectors used in CAR-T cell therapyWith the increasing progress in genetic modification technologies, several approaches have already been tested to generate CAR-T cells. The two main methods are viral transduction (retroviral or lentiviral vector), or transfection with naked plasmid DNA, transposase DNA-mediated integration or mRNA by electroporation or by lipid-based transfection system.3 Commonly used vectors with their advantages and disadvantages are listed in Table 1. Most current studies use retroviral vectors (such as lenti- and retroviral vectors). Lentiviral vectors have become particularly attractive for clinical applications due to their ability to efficiently transduce most cell types, including non-proliferating cells, such as Naive T cells.4 Both γ-retroviruses and lentiviruses integrate semi-randomly into the host cell genome. Thus, presenting a risk of insertional mutagenesis and dysregulation of genes adjacent to the integration site5 and local hotspots.6
Vectors currently used in CAR-T cell therapy.
IP: intellectual property.
The development and use of non-integrative vectors, or ones that integrate at specific locations, is an important goal for CAR therapy. The development of new vectors has been driven mainly by the need to address the safety issue, in particular the problem of insertional mutagenesis.7
Non-viral vectors such as Sleeping Beauty (SB), piggyBac (PB) or Tol2 derived transposons typically require co-transfection of the transposon DNA with a transposase such as an expression plasmid or mRNA. Consequently, this results in genomic integration generally into AT-rich genomic regions.8 Although the integration profile of transposon can be considered biologically safe, recently two patients developed malignant lymphoma after a treatment with CAR-T cells anti CD19 using a PB vector.9 In addition, they are easier to produce on a large scale. In addition, they harbor greater transgenic capacity and generate fewer concerns about biosafety. However, an important difference between the various technologies is the duration of CAR expression in the modified cell. For longer expression (multiple weeks), viral transduction is usually employed, while mRNA electroporation results in transient expression lasting for about one-two weeks.
The affinity of the scFv can impact CAR specificity and off target potentialMost of the molecular targets for CAR-T cells are not exclusively expressed in tumor cells. Some of these molecules can be found expressed at different levels in tumor and healthy cells. Exploring scFvs recognizing the target with different affinities in CAR molecules can potentially allow the discrimination between healthy and tumor cells. Some groups have explored such approach, especially in the preclinical setting10 and reviewed in.11 Recent results showed potential toxicity due to on-target off-tumor recognition when using high affinity CARs,12 and this also advocates in favor of reduced CAR affinities in specific contexts. Reducing the affinity of anti CD19 scFvs has also shown potential benefits, such as delayed kinetics of T cell activation (and potentially less induction of CRS) without loss of tumor elimination capacity.13 One critical aspect of this approach is that some target antigens can be modulated in different cell types according to tissue inflammation status of pharmacological exposure. As an example, CD22, an ALL-target antigen, can be modulated with drugs both in vitro and in vivo, augmenting the recognition and killing by CD22-specific CARs.14 Such contexts make it challenging to fine tune target recognition based on the balance of scFv affinity and target molecule expression density.
Characterization and control testing of vectors productionDNA vectors productionWhether using viral or non-viral vectors, the start of production of most vectors starts with the production of plasmid DNA. Testing of DNA vectors should include tests for genetic identity and integrity including confirmation of the therapeutic sequence and regulatory/controlling sequences, freedom from extraneous agents, sterility and endotoxin levels. Also, the presence/absence of specific features such as CpG sequences should be confirmed by suitable methods.15
Cell banksEukaryotic cell bank tests (master and working banks) conducted on producer/packer cell lines should include identity, purity, cell number, viability, strain characterization, genotyping/phenotyping. In addition, tests must be performed to verify possible contamination with adventitious viruses, absence of bacterial and fungal contamination, as well as mycoplasma.16
Viral vectorsAny preparation of viral vectors should be fully characterized with regard to transducing activity, other characteristics relevant to vector particles and the absence of RCL (replication competent lentivirus) or RCR (replication competent retrovirus). Lot-release specifications should be based on appropriate tests to characterize and assure the integrity of the vector. For any viral vector the maximal level of contamination by plasmid DNA in the final lot should be set and it is recommended DNase treatment to remove plasmid DNA. DNAse treatment is essential to eliminate possible contaminations in the viral product with plasmid encoding VSV-G, other envelope proteins and other viral proteins such as Gag/Pol.16
Retroviral vectors are being used in a growing number of CAR-T clinical applications. As the use of this type of vector is increasing, a safety concern about a possible viral recombination and the risk of developing a replication competent retrovirus (RCR) during manufacture of the vector material. The potential pathogenicity of replication competent retrovirus (RCR) requires vigilant testing to exclude the presence of RCR in retroviral vector-based human gene therapy products. Lentivirus RCR is referred to as replication competent lentivirus (RCL). To manage this risk, regulators have required screening of T cell products infused into patients for RCR/RCL.17,18
Transducing activityImportant aspects of transducing activity are integration capacity, transgene expression and functionality. There are two common measurement types: physical, which determine particle concentration, e.g., p24 quantification or functional, which count particles capable of productive transduction (IFU/ml). Productive production can be measured by using limiting dilution techniques of the virus to transduce a specific number of cells and the transduction activity can be measured by flow cytometry or by qPCR.19,20
Characterization and control testing of CAR-T cells productionSerological testsUsually, patients undergoing CAR-T cell therapy are repeatedly hospitalized and exposed to blood products. Therefore, testing for infectious agents should be performed as a quality control step in order to ensure the biosafety of the source material. The minimal suggested panel for infectious diseases prior to CAR-T cell manufacturing is the following:
- •
Anti HTLV I and HTLV II (HTLV I/II);
- •
CMV antibody;
- •
Hepatitis B Antigen (HBSAG);
- •
Hepatitis B core Antibody (HBCAB);
- •
Serology test for syphilis (RPR);
- •
HIV/HCV antigen, performed by Nucleic Acid Testing (NAT);
- •
HIV by NAT;
- •
HCV by NAT;
- •
Anti-HIV-1 and HIV-2 (HIV 1/2);
- •
Hepatitis C Virus Antibody (HCVAB or HCV).
The expected results should be HIV/HCV by NAT, Anti-HIV1/2, HTLV I/II, HBSAG, HBCAB, HCVAB (HCV), and RPR = Non-Reactive (NR). CMV may be either positive or negative and is tested for information only.
CytogeneticsThe extensive time of culture and expansion of T cells requires genomic integrity tests in order to ensure that no aberrant clones would be generated as a consequence of viral integration nor deletions/duplications of genes or larger chromosomal bands occur. Usually, the analysis of at least 20 metaphases in a G-band karyotype is the minimal informative test to check for chromosomal abnormalities or aneuploidies. However, karyotype tests are not informative for deletions or duplications at a molecular level. If a more accurate screening is necessary a genomic array must be considered for a genome-wide screening of copy-number abnormalities and the detection of loss of heterozygosity.21
Sterility testsOnce the production of CAR-T cells involves extensive product handling with a diversity of reagents and plastic consumables and fully closed systems are not always available, sterility tests are mandatory to check for the presence of microbiological contaminants before infusion. Basically, the routine tests screen for mycoplasma, bacterial, fungal and endotoxin contamination.22 All tests should preferably be performed with kits approved by the competent regulatory agencies. Any contaminated products should not be used for infusion under any circumstance.
Mycoplasma tests usually involve two different techniques: 1) The use of bioluminescence to check for the presence of mycoplasma enzymes in the final product or 2) a PCR test with oligos that amplify specific regions of the mycoplasma genome. Both approaches use a small sample of the final product's supernatant to extract the mycoplasma cytoplasmic/genetic material. The performance of these two techniques is comparable, however in-house validation is needed.22
Bacterial and Fungal contamination can be assessed through automated culture methods and might be performed both prior and after CAR-T cell production. The most common methods detect CO2 production by microorganisms displaying the results by changes in the culture media color or in the bottle pressure. The main panels of pathogens include S. aureus, P. aeruginosa, B. subtilis, C. sporogenes and C. albicans.23
Finally, bacterial endotoxin testing (BET) aims to detect the presence of substances released by lysed bacteria that might cause toxic effects (e.g. gram-negative LPS). The most popular BET is the Limulus Amebocyte Lysate (LAL) which is based on the reactivity of seahorse amebocytes when exposed to endotoxins. The four main types of LAL test include one qualitative (gel clot) and three quantitative assays (turbidimetric, chromogenic and recombinant Factor C). The indicated endotoxin levels in the final product need to be below 5 EU/kg.24
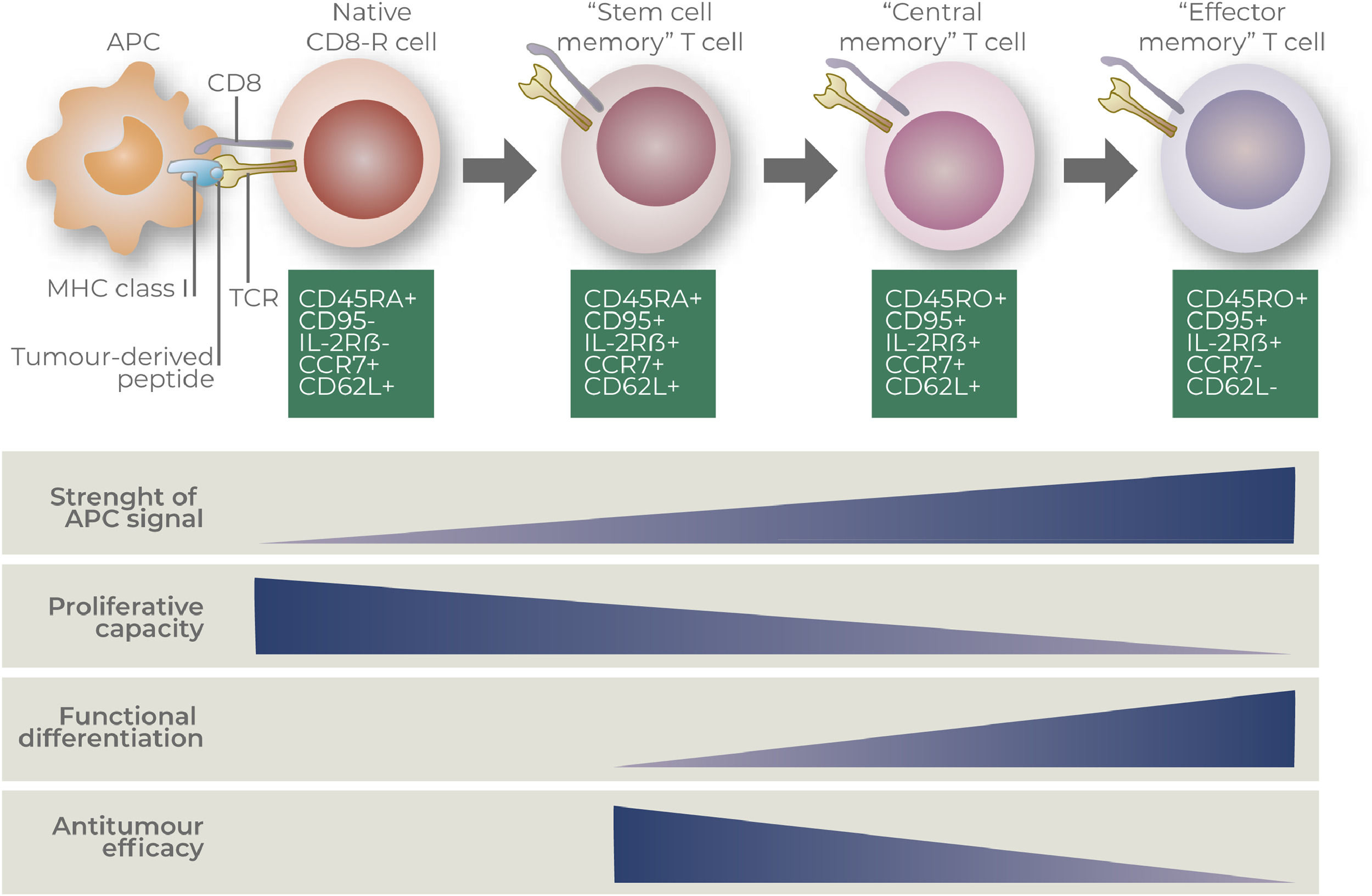
Phenotypic and functional characterization of CAR-T cell productsMolecular characterizationCAR-T cell products are generally evaluated according to its memory-effector phenotype via transcriptomic and/or proteomic analysis. Most common techniques used to characterize CAR-T cells may include RNA-sequencing, single-cell RNA-sequencing and multiparametric flow cytometry.25–27 Transcription factors and surface molecules may also define the status of T cell differentiation, which may correspond to their ability to respond to tumor-antigens, self-renew, migrate, proliferate and become exhausted.28–30 Most common used markers to define T cell subpopulations are based on the expression of surface markers CD45RA, CD45RO, CCR7, CD62L, CD95 and CD27.31–33 Phenotypic characteristics and functional features of distinct CD8+ T cells subsets are detailed at Figure 2, according to Gattinoni and collaborators.34
.34")
Description of CD8+ T cell states of differentiation (Gattinoni and collaborators).34
The evaluation of cytokine production represents an important step to the profiling of the final CAR-T cell products. The release of cytokines may provide functional insights concerning the status of differentiation (e.g. effector, memory, stemness profiles), the spectrum of polyfunctionality acquired, as well as the amplitude and potency of T cell responses, taking into account also its neurotoxic potential.35
The most applied tests to evaluate cytokine production include the enzyme-linked immunosorbent assay (ELISA), the enzyme-linked immunospot (ELISpot),36 intracellular cytokine staining by flow and newly described multiplexed techniques to evaluate the compounds at the single-cell level.37 The production of cytokines represents an important assay for the characterization of functional CAR-T cell products, however there are no guidelines defining this step as a validation requirement for infusion.
A series of studies have used a standard panel of cytokines for the classical characterization of T cell activation, including IFN-gamma, TNF-alpha, Granzyme-B among the evaluated factors.31,38–40 The pattern of cytokine production may differ according to the choice of the original cell type for CAR-T cell manufacturing. The use of whole PBMCs or total CD3+ T cells in the first step of cell selection may drive the outline of cytokines/chemokines towards a mixed phenotype, including also cytokines typically produced by CD4+ T helper cells, such as IL-2, IL-4, IL-5, IL-10, IL-13, IL-17. In contrast, CAR-T cells generated from isolated CD8+ T cells will mostly produce IFN-gamma, TNF-alpha, perforin and granzyme-B upon stimulation.
Importantly, recent findings by Alizadeh and collaborators41 have described an important role of IFN-gamma producing-CAR-T cells not only in eliminating tumor cells and tumor control, but also in the systemic stimulation of innate immunity from the host, by stimulating macrophage proinflammatory functions. Some additional studies have concentrated their efforts to generate CAR-T cells that acquire polyfunctional features. These cells are normally defined as being able to secrete at least two cytokines simultaneously, which represents a great gain in the stimulatory capability in vivo.42 The generation of polyfunctional CAR-T cells were described in pre-clinical models43 and Phase-I/II trials for non-Hodgkin Lymphoma.44,45 In fact, polyfunctional T cells have been largely associated to a better prognosis for distinct solid and hematologic cancer types,46–48 but improvements in protocols for the activation and expansion of CAR-T cells are needed to generate a sustained maintenance of these cells in vivo.
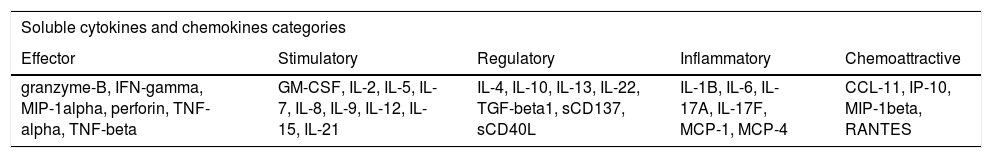
Distinct groups of cytokines and chemokines could be evaluated for CAR-T cell profiling. Molecules can be subdivided into five categories, accordingly to Rossi and colleagues44: effector, stimulatory, regulatory, inflammatory and chemoattractive (Table 2).
Description of cytokines and chemokines for CAR-T cells profiling.
GM-CSF: Granulocyte-macrophage colony-stimulating factor; IL: Interleukin; TGF-Beta1: Transforming growth factor beta 1; MCP: Monocyte chemoattractant protein; CCL11: C-C Motif chemokine ligand 11; IP-10: Interferon gamma-induced protein 10; MIP: Macrophage inflammatory protein; RANTES: Regulated upon Activation, Normal T-cell Expressed, and Secreted.
By using cytokine single-cell approaches Rossi and collaborators44 have reported that about 20% of CAR-T cells products presented a polyfunctional profiling in their protocol. Curiously, patients showing objective responses were infused with polyfunctional CD4+ CAR-T cells that secreted inflammatory cytokines, regulatory cytokines and effector, while non-responding patients received polyfunctional CD4+ CAR-T cells that produced mostly effector molecules, suggesting that grater polyfunctional features of CAR-T cell may represent a benefit for patients.
Co-culture assaysThe functionality of CAR-T cell products is based mostly on its ability to effectively recognize and destroy target neoplastic cells.49 Even considering that some CAR-T cell preparations may include expanded-CD4+ T cells, the great majority of assays are focused on the role of cytotoxic CD8+ T lymphocytes (CTLs) in the elimination of tumor cells. CAR-T cell specificity is HLA independent and relies mostly on the targeting capability of its extracellular binding domain to cell surface antigens, including proteins, carbohydrates and glycolipids.50 The classically described mechanisms comprise the Fas/Fas ligand (FasL) interaction pathway, perforin/granzyme secretion and the production of effector cytokines such as IFN-gamma and TNF-alpha.51 In order to evaluate the cytotoxic effect of the CAR-T cells, standard co-culture assays were described.52
Following antigen recognition, the killing activated T cells can be determined by a cytokine release assay, and in vitro cytokine production and cytolytic activity of CD8+ T cells are often correlated, mainly because IFN-γ enhance MHC-I and Fas expression on target cells.53,54 On the other hand, detection of cytokine release can only be considered as a potential marker for cytolytic function, because killing of target cells requires lymphocyte production and subsequent release of mediators for death induction, as well as cell-cell interaction.55
Number of integrated copiesThere is an increased risk of oncogenesis if the vector copy number (VCN) per cell is high. Due to the wide distribution of proviral DNA insertion sites, the risk of oncogenesis due to insertional mutagenesis increases with the number of modified cells and with the number of insertions per cellular genome. For this reason, it is highly recommended the VCN shall be <5 copies per genome.16 Accurate and rapid measurement of VCN is an important quality control step required for release of CAR T-cell products for patient infusion and the identification of the integration sites in the human genome may contribute to safety assurance.
In vivo assays - methodologies and animal modelsMouse models have been of pivotal importance in determining the efficacy and safety of CAR-T cells therapy. According to the nature of the tumor graft and of its recipient, mouse models can be categorized into four groups: a) syngeneic; b) human xenografts; c) immunocompetent transgenic, and d) humanized transgenic mouse models.56
Syngeneic or immunocompetent allograft mouse models use CAR‐T cells, tumors, and target antigens that are all murine‐derived. The high ground of this model is that the recipient has a fully physiologic immune system, thus representing an excellent tool to observe CAR‐T cells' interaction with other cells and elements of the immune system. Additionally, syngeneic models can reveal toxicities caused by the presence of tumor-associated antigens (TAAs) in healthy murine tissues, the so-called on-target off‐tumor toxicities. Nevertheless, as mouse biology does not always accurately recapitulate human biology and many important adverse events associated with CAR-T cells therapy were not observed in the syngeneic models, namely the cytokine release syndrome (CRS). In addition, the persistence in the circulation of murine CAR‐T cells is shorter compared to human CAR‐T cells.57 An important example of the contribution of the syngeneic models is provided by the studies of David Gilham's group showing that second-generation CARs with CD28 costimulatory domains induced B cell aplasia and chronic toxicity accompanied by an increase in CD11b+Gr‐1+ myeloid‐derived suppressor cells (MDSCs).58,59 The unexpected effect on MDSCs could be detected only in a syngeneic model.
The xenograft models are based on the injection of human tumors and CAR‐T cells into an immunocompromised mouse. Currently, most xenograft mouse models of CAR‐T therapy use the NOD‐SCID‐IL2rγnull (NSG) mouse strain.60 The parental NOD-SCID strain was obtained by crossing non‐obese diabetic (NOD) mice, which have impaired innate immunity, with severe combined immunodeficiency (SCID) mice, which are deficient in the adaptive immune system. NSG mice were developed by introducing a mutation in the IL‐2 receptor γ chain gene, which resulted in the loss of signal transduction from multiple cytokines and increased immunodeficiency compared to parental NOD-SCID mice. The xenograft models were frequently used to validate proof-of-concept studies. As in this model, human CAR-T cells interact with human tumoral tissue in the absence of an intact immune system, and xenograft models are tools to test the selectivity of more complex or novel CAR constructs. One area of interest is in developing CARs that are dependent on multiple TAAs for full activation to decrease the chance of off‐tumor effects or antigen escape. Examples of the new strategies of CAR-T cells evaluated in xenograft models are a) bispecific CARs targeting both CD19 and CD20 to treat lymphomas and acute lymphoblastic leukemias,61 b) masked anti‐EGFR CARs in which the scFVs were masked by a peptide with a linker that is cleavable by proteases expressed by tumor cells but not healthy tissues62 c) Switch-mediated activation and retargeting of CAR-T cells that targeted a specific peptide neo‐epitope introduced on a TAA‐binding antibody.63 The xenograft models were also used to understand the mechanisms of CAR-T cells exhaustion. In two second-generation human CAR‐T cells, exhaustion was prevented with 4‐1BB costimulation and exacerbated by CD28 costimulation.64
Patient‐derived xenograft (PDX) models are a specific subtype of xenograft model generated by implanting a primary tumor biopsy instead of injecting tumor cell lines in an immunocompromised host. The advantages of PDX models are to better capture tumor heterogeneity and to avoid exposure to artificial environments used in the in vitro experiments. In a hepatocarcinoma PDX model, Jiang et al. showed that it accurately predicted patient response and could be useful in determining the course of treatment, such as a combination of checkpoint blockade and CAR‐T therapies.65
Immunocompetent transgenic mice have been the less frequently used model employed in CAR‐T studies. Their utility relies on their potential for the evaluation of on‐target off‐tumor effects. These transgenic mice are engineered to lack the expression of a murine TAA (knockout) and to express a human TAA (knockin) in specific tissues and at a certain level. Pegram et al. reported that treatment with CD19-specific, CAR-T cells that were further modified to constitutively secrete IL-12 were able to eradicate established disease in the absence of prior conditioning.38 Moreover, the tumor elimination required both CD4(+) and CD8(+) T-cell subsets, autocrine IL-12 stimulation, and subsequent IFNγ secretion by the CAR(+) T cells. The authors proposed that adoptive therapy using CAR-targeted T cells modified to secrete IL-12 would obviate or reduce the need for conditioning regimens.38
Humanized transgenic mice are immunocompromised mice implanted with human immune cells in addition to human tumor and CAR‐T cells. The main advantage of this model is the fact that mice are tolerant to human cells yet have aspects of a human immune system. Several groups have used NSG mice transplanted with human CD34+ cells to look for CAR‐T toxicity against hematopoietic stem cells (HSPCs). In the humanized immune system (HIS) model, sublethally irradiated newborn BALB/c Rag2‐/‐γc ‐/‐ mice are injected with human CD34+ cells. The use of newborn immunodeficient mice was important because they support better T cell development, as they do not display the thymic involution and phagocytic activity against human immune cell engraftment seen in adult mice. Hombach et al. used a HIS model to test if anti‐CD30 CAR‐T cells could eradicate lymphoma without lasting B cell aplasia.66 CD30 is also expressed by HSPCs during activation but in lower levels than in lymphoma cells. This difference in expression level, as well as the expression of granzyme B inhibitor by HSPCs, may have contributed to the protection of the engrafted HSPCs from CAR‐T‐mediated toxicity. A major concern in humanized models is the fact that the rates of lymphoid and myeloid cells produced by human CD34+ cells, as well as the profile of the circulating cytokines, are distinct from those found in humans, and therefore do not reflect the human immune cell development.
From the above, it is clear that there is no single mouse model capable of encompassing all aspects involved in the efficacy and safety for CAR‐T therapy, but this is a moving field and there has been significant improvements in the humanization of murine immune systems. At present, we still need the association of multiple animal models that will provide complementary information of different aspects of CAR‐T treatment. Nevertheless, it is evident that animal models are essential tools to predict CAR‐T safety and efficacy in the clinic.
ConclusionThe science of CAR-T cell development and production has evolved at a fast pace in recent years. Although many biological aspects of CAR expressing cells are still being uncovered, minimal parameters for CAR-T cell function characterization are now widely accepted by the scientific community. We approached herein most of the important aspects for characterizing a functional and high-quality CAR-based cell product. The biological assays for evaluating such parameters are also impacted by new technological developments, and regular updates for the validation tests are expected.
It is important to note that new technologies are being developed and disruptive science is supporting CAR-T cell developments, so new aspects of CAR-T cell biology, such as surrogate markers for in vivo antitumor effectivity and exhaustion/dysfunction are likely to be incorporated as evaluation criteria in the near future.
RLGC is supported by CNPq# 442686/2020-0, Pronon# 25000.021774/2019-13 and ASH Global Research Award. VPC is supported by FAPESP# 2019/25309-0, FAPESP# 2013/08135-2, CNPq# 442484/2020-8 and Pronon# 25000.189625/2016-16. MHB is supported by grants of CNPq and FAPERJ. EMR and VCP are also supported by FAPESP# 2020/07055-9. RNR and TGMO are supported by CNPq# 44676/2020-4.


